
Epigenetics, a key player of immunotherapy resistance


Abstract
In 2018, the Nobel Prize in medicine was awarded to James P. Allison and Tasuku Honjo for their work on the description of immune checkpoint inhibitors which contributed to the development of new anti-cancer immunotherapies. However, although these new therapeutic strategies, which are designed to limit immune escape of cancer cells, have been used or tested successfully in many different cancers, a large proportion of patients have been described to resist and not respond to these new treatments. The new incoming challenge is now therefore to overcome these resistance and new recent data presented epigenetic modifications as promising targets to restore anti-tumor immunity. Indeed, both DNA methylation and post-translational histone modifications have been described to regulate immune checkpoint inhibitor expression, tumor-associated antigen presentation or cancer cell editing by the immune system and therefore establishing epigenetic drugs as a potential complement to immunotherapies to improve their efficiency.
Keywords
Introduction
Antigen presentation by the professional antigen-presenting cells (APCs) are normally recognized by immune cells via a surface T-cell receptor (TCR), leading to the induction of naive CD8+ T-cell clonal expansion and differentiation and finally resulting in the cell death of APCs. Specific tumor-associated antigens (TAAs) expressed by cancer cells can be identified by the immune system and results in the activation of immune system effectors and the consequent elimination of the cancer cells. Indeed, an increase in the recruitment of cytotoxic CD8+ T-lymphocytes into the tumor is mediated by chemokines such as C-C motif chemokine ligand 5, C-X-C motif chemokine ligand 9 (CXCL9) and CXCL10 and is correlated to lower metastasis risk and better outcome for patients[1-3][Figure 1]. Three sequential phases of immune edition of cancers have been described and summarized under the rule of the “3E”: (1) Elimination; (2) Equilibrium; and (3) Escape[4]. During the Elimination phase, some newly transformed cells can be quickly eliminated by immune effector cells, such as natural killers (NKs), but this phase can also favor the development of heterogeneous tumor cancer cells resulting in the selection of new variants resistant to immune edition. Further growth of cancer cells selected during the Equilibrium phase may then lead to the acquisition of aggressive phenotypes and resistance to immune system elimination inducing clinical lesions. So, immune escape has now been included as one of the new hallmarks of cancer development described in the revised version of Hanahan and Weinberg[5] in 2011, together with resistance to cell death, replicative immortality or induced angiogenesis, for example.
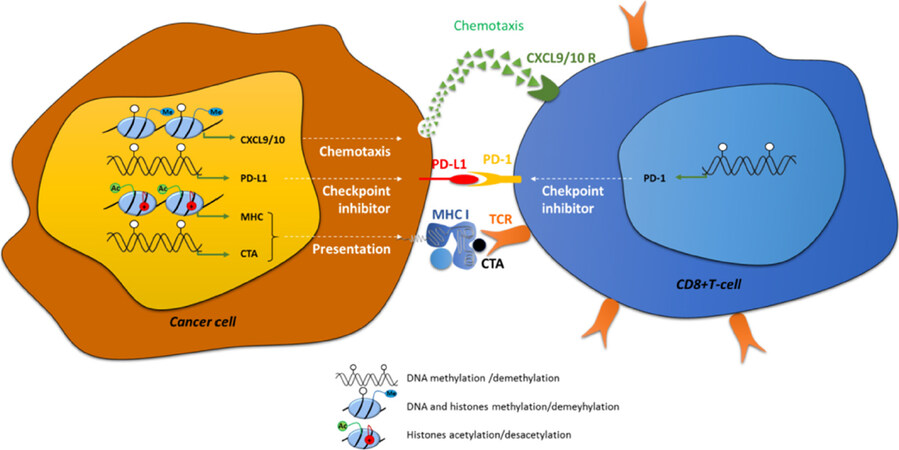
Figure 1. Examples of direct implication of epigenetic modifications in immune recognition of cancer cells. Epigenetic modifications: DNA methylation (white circles) histone methylation (me) and histone acetylation (Ac, +) are directly implicated in: (1) chemotaxis by the regulation of the expression of CXCL9/10; (2) in immune checkpoint control, by the regulation of PD-L1 and PD-1 in cancer cells and CD8+ cells respectively; and in (3) antigen presentation, by the control of the expression of CTAs and MHC-I. CXCL: C-X-C motif chemokine ligand; PD-1: programed cell death 1; PD-L1: PD-1 ligand; CTA: cancer testis antigen; TCR: T-cell receptor
Resistance to immunotherapy in cancers
Immunotherapy protocols are designed to annihilate immune escape in cancer cells and restore elimination of cancer cells. In 2018, Nobel Prize in medicine awarded James P. Allison and Tasuku Honjo for their work on the knowledge of immune edition and the development of immunotherapies to fight against cancer cells[6,7]. Immunotherapies include: cancer vaccines, humanized monoclonal antibodies targeting TAAs, transfer of chimeric antigen receptor-T-cells or adoptive transfer of transgenic TCR-expressing T-cells. Nevertheless, immunotherapy resistances have been described in patients and are divided in innate and acquired resistance (for a review[8]). Intrinsic resistance is frequently observed in patients with a failure of systemic immune response such as immune-compromised HIV patients or specific failures induced for example by a lack of expression of specific tumour antigens by tumour cells. Adaptive resistance might be provoked by several processes [Figure 1]: (1) loss of antigens presentation due to mysfunctions in antigen presentation machinery or a decrease in major histocompatibility complex (MHC) expression; (2) dysfunctions of signaling pathways such as the Wnt/βcatenin, phosphatase and tensin homolog, phosphoinositide-3-kinase (PI3K) or Janus kinase pathways[9-12]; and (3) prolonged T-cell stimulation, which is frequently observed in chronic diseases such as chronic infection or cancer. This stimulation frequently leads to the exhaustion of immune cells, phenotype which is characterized by a loss of both immune and proliferative capacities. This state is usually accompanied by a loss of expression of cytokines such as interleukin-2 (IL-2), tumor necrosis factor α (TNFα) and interferon (IFN) and induction of immune checkpoint inhibitors such as programed cell death 1 (PD-1) (PDCD1), cytotoxic T-lymphocyte associated protein 4 (CTLA-4), lymphocyte activating 3 (LAG-3) and T-cell immunoglobulin mucin 3 (TIM-3)[13]. Immune checkpoint inhibitors are proteins controlling the immune system: PD-1 is expressed at the surface of various immune cells, such as T-lymphocytes, myeloid cells or NKs, and is particularly expressed in exhausted T-cells. PD-1 ligand (PD-L1) (also referred as CD274) or PD-L2 are expressed at the cell surface of tumor cells and can bind to the PD-1 receptor [Figure 1]. The PD-1/PD-L1/2 interaction induces a negative signaling cascade which leads to the inhibition of CD8+ T-cell proliferation, cytokine secretion and inflammation leading to a decrease in tumor cell elimination. Moreover, interaction of the other immune check point inhibitors with their respective ligands (CTLA-4 with the ligands CD80/CD86 expressed on regulator T-cells, LAG-3 with MHC class II, LAG-3 with galectin-9, high mobility group protein B1 or with carcinoembryonic antigen cell adhesion molecule 1 and phosphatidylserine) also decrease the anti-tumour immune response.
Although the identification of these mechanisms is recent and most of the factors involved still remain largely unknown. Anti-PD-1 and anti-PD-L1 immunotherapy protocols (such as nivolumab or pembrolizumad) have been developed and used to restore immune edition in cancers [Figure 2]. This process is called immune checkpoint blockade (ICB) and, in the last few years, this new treatment has been tested and shown promising results in many different types of cancers such as: non-small cell lung carcinoma (NSCLC), melanoma, mesothelioma, renal cell carcinoma (RCC), bladder cancer, head and neck squamous cell carcinoma (HNSCC)[14-19]. Unfortunately, although some very interesting results were obtained in some patients, resistance to ICB are observed in a large percentage of cases. For example, only 20% of NSCLC or triple negative breast cancer (TNBC) patients treated with an anti-PD-1 therapy presented a significant positive response[20,21]. These results suggest that immunotherapy resistance is present in a large proportion of patients before treatment and may be due to a low immune checkpoint inhibitor expression or an absence of T-cell infiltration in the solid tumors.
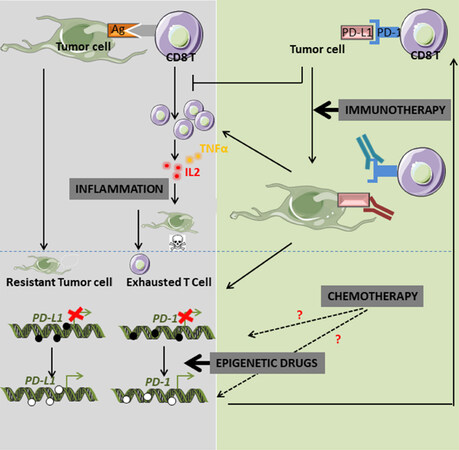
Figure 2. Crosstalk between epigenetics, PD-L1/PD-L1 expression and immunotherapy. PD-1: programed cell death 1; PD-L1: PD-1 ligand
Immune checkpoint inhibitors are epigenetically controlled
Epigenetics includes DNA methylation, histones post-translational modifications and non-coding RNA and regulates gene expression in a transmissible but reversible manner. DNA methylation is processed by DNA methyl transferases (DNMTs) which catalyze the addition of a 5methyl cytosine on the 5th position of cytosines in CpGs. This mark is generally repressive and many genes are normally controlled by DNA methylation during embryogenesis. Aberrant DNA methylation also frequently occurs in pathologies, specifically in cancers, where it contributes to tumor suppressor gene silencing or inactivation of apoptosis (for a review[22]). Histones are organized in nucleosomes whose local compaction is regulated by histones post-translational modifications. These modifications are varied but the most studied are acetylation and methylation. Acetylation on lysines in histone tails is promoted by histone acetyl transferases and provokes a relaxed chromatin favorable to transcription factor recruitment, opposite to deacetylation which is catalyzed by histones deacetylases (HDACs). The pro- or anti-transcriptional role of histone lysine and arginine methylation is dependent of both the position of the amino acid and the level of methylation. For example, the H3K4me2/3 mark is permissive whereas the H3K9me2/3 or H3K27me2/3 are repressive. These modifications are catalyzed by histone methyl transferases (HMTs) and removed by histone demethylases. In regard to their roles in tumorigenesis, the epigenetic enzymes are potential targets for anti-tumoral therapies used routinely in some specific cancers or tested in clinical trials (for review[23]).
Although the regulation of expression of PD-1 and PD-L1 in tumors is still largely unknown, recent data proposed a major involvement of epigenetics. Wrangle and collaborators were the first to demonstrate, using genome-wide studies in NSCLC cell lines, that the passive DNA-demethylating agent azacytidine (Aza) potentiated innate and adaptive immune pathways. This effect was associated with an increase in PD-L1 expression[24]. They first observed in a phase I study that combination of Aza with entinostat induced a significant objective response to classical anticancer agents in a small percentage of patients (4 out of 19 NSCLCs)[25]. They then theorized that epigenetic drugs might restore sensitivity to anti-PD-1 therapy into specific patients. Epigenetic modifications are crucial for immunotherapy resistance, not only by controlling immune checkpoint inhibitor expression, but also by regulating immune cells infiltration within the tumor, antigen presentation or cytokine profile expression and secretion [Figure 1]. Indeed, increased recruitment of CD8+ tumor-infiltrating lymphocytes (TILs) are directly linked to clinical outcome in numerous cancers, such as breast cancer or ovarian cancer[26,27]. It is now described that this infiltrate is regulated by epigenetic modifications. For example, the HMT enhancer of zeste homolog 2, which mediates the trimethylation of histone H3 lysine 27 (H3K27me3), and DNMT1 which repressed the production and secretion of CXCL9 and CXCL10 by Th1 therefore inhibited the recruitment of CD8+ T-cells within the tumor in ovarian cancer models[1]. Moreover, decitabine was also found to increase CD80 expression on tumor cells, inducing T lymphocytes infiltration in mouse EL4 tumor model and tumor rejection[28].
Increased expression of PD-1 in breast cancers and colorectal carcinoma was related to both decreased DNA methylation and decreased repressive H3K9me3 and or H3K27me3 histone marks on the PD-1 promoter[29-31]. However, no changes in epigenetic modifications were observed on the PD-L1 promoter in these cancers, probably due to its unmethylated status in normal tissues. Similarly, the expression of the immune checkpoint inhibitors CTLA-4, TIM-3 and LAG-3 were also up-regulated in breast cancers compared to normal adjacent tissues and their promoters presented lower DNA methylation and lower repressive histone marks[30]. Only one exception was described since the decreased expression of the immune checkpoint inhibitor LAG-3 appeared to be specifically associated to variations of histone modifications (H3K9me3 and H3K27me3)[29,32]. Similar results showed an inverse correlation between promoter DNA methylation of PD-1, PD-L1, CTLA4 and expression of these proteins in NSCLCs compared to normal tissues[33,34]. Moreover, methylation of the CTLA4 promoter was also inversely correlated to its expression in a large cohort (470) of malignant melanoma patients treated with anti-PD-1/PD-L1 antibodies. Furthermore, a strong significant correlation was observed between low promoter methylation of CTLA4 and both positive response to immunotherapy and survival[35]. In prostate cancers, methylation of PD-1 promoter was more frequently observed than in normal tissues and its expression was inversely correlated to its promoter methylation[36]. Moreover, patients presenting a high level of methylated PD-1 promoter presented a higher risk of recurrence in prostate cancer. In leukemia, PD-L1 expression has also been associated with low promoter methylation and to poor prognosis[37]. The demethylation of an enhancer region of PD-1 and the protein re-expression was reported in CD8+ T-cells in chronic lymphocytic leukemia[38]. In diffuse low grade glioma (LGG), methylation of PD-L1, PD-L2 and CTLA-4 promoters was inversely correlated to their expression. We also recently reported in epithelial to mesenchymal transition-induced lung cancer cell models (A549) that transforming growth factor β (TGFβ), a cytokine frequently expressed in the tumor microenvironment, was responsible for the demethylation of the PD-L1 promoter by inhibiting both global and local DNA methylation[39]. In addition, the combined treatment of these cells with TNFα favored NFκB signaling and PD-L1 transcription by inducing the recruitment of NFκB/p65 on the PD-L1 promoter. Besides the tumor microenvironment which could severely affect PD-1/PD-L1 expression, cancer drugs might also modify their expression, thus inducing drug resistance. For example, 30% of melanoma patients treated with ICB who presented a positive response towards the protocol, developed secondary tumors (for a review[40]). Exhausted T-cells which appeared during ICB are supposed to present an epigenetic-linked genome wide regulation mediated by the de novo DNMT3A leading to the inhibition of proliferation and metabolic activity and thus restricting clonal CD8+ T-cell diversity. Indeed, DNA methylation inhibitors improved T-cell response in anti-PD-1 therapy[41]. On the opposite, an acquired resistance was frequently found in myelodysplastic syndrome (MDS) in which the use of DNA hypomethylating agents is the standard treatment. Inhibition of DNA methylation induces an increase in PD-1 in T-cells of these patients[42]. Although mechanisms of resistance to demethylating agents are still unclear, it has been suggested that the activation of the immune checkpoint inhibitor was more likely involved. Moreover, treatment of NSCLC patients with conventional chemotherapy or epidermal growth factor receptor inhibitors increased PD-L1 expression unlike anti-PD-L1 treatments which significantly decreased PD-L1 expression[34]. In addition, an anti-PD-L1 treatment of primary tumors which expressed low levels of PD-L1, induced secondary tumours presenting an increase in the methylation of PD-L1 promoter.
A recent multi-country (France, Spain and Italy) cohort study of 142 high grade NSCLC samples highlighted a DNA methylation specific signature which can significantly predict which patients could positively benefit from nivolumab or pembrolizumad treatments[43]. DNA repair pathways have been associated with efficiency of ICB treatments. Indeed, a study analyzing the methylation status of 179 genes involved in DNA repair in HNSCC, cervical carcinoma and laryngeal squamous cell carcinoma identified 15, 3 and 2 genes, respectively, whose methylation was associated with increased PD-L1 expression. Amongst these genes, RAD51B was found in the three different cancer models suggesting that alteration of homologous DNA reparation strongly favored immune checkpoint inhibitor expression[44].
Only 15% of metastatic HNSCC presented an objective response to anti-PD-1/PD-L1 treatments. In these cancers, both PD-L1 and PD-L2 expression was increased in tumor cells compared to normal adjacent tissues and their expression was inversely correlated to methylation of their promoters[45]. Interestingly, human papillomavirus-positive HNSCC were strongly associated with both PD-1 and PD-L1 promoter methylation and poor prognosis suggesting that these particular tumors are unlikely to respond to immunotherapy[45,46]. Treatment of mesothelioma cells using the demethylating agent decitabine alone is unable to significantly restore PD-L1 expression but combination of decitabine with HDACi [valproic acid, suberoylanilide hydroxamic acid (SAHA) or NODH] strongly induced PD-L1 transcription in these cells[30]. Expressions of PD-L1 and CTLA4 mRNA in acute myeloid leukemia (AML) patients treated with combined HDACi and DNMTi were also increased during treatment but presented very high oscillations. Interestingly, decitabine alone was sufficient to restore both mRNA and protein PD-L1, PD-1 and CTLA4 expression in a dose-dependent manner in AML cell lines[47].
Combination of epidrugs with immunotherapies restores treatment efficiency
Cell models
Although some studies showed a positive effect of 5-Aza treatment on PD-L1 expression in NSCLC cell lines, only 13% of NSCLC tumors issued from patients treated with this demethylating agent, presented a strong expression of PD-L1 protein (however, PD-L1 expression was at least detectable in 55% of cases). These data strongly support the hypothesis that different cellular mechanisms control PD-L1 expression. Indeed, the competitive inhibitor of DNMT1, procainamide or 5-Aza treatments alone failed to induce PD-L1 expression in the NSLCC cell line A549 whereas the combination of these compounds with TNFα reactivated PD-L1 expression[39]. In NSCLC models (A549 and H838 cells), the DNA methylation inhibitor Aza, induced the demethylation of promoters, increased the expression of interferon regulating factor 1 (IRF1) and provoked an increase in PD-L1 expression in both a interferon-dependent and independent manner[48]. Moreover, expression of CXCL9 and CXCL10, consecutive to the IRFs activation, improved the efficiency of anti-PD-L1 protocols in mice models.
Pre-clinical studies
Despite obtaining better therapeutic responses with combined molecules than single drug treatment, combined anti-PD-1 and anti-CTLA4 therapy could not limit the formation and tumor growth of CT26 and 4T1 cancer models in mice[49]. However, HDACi significantly increased PD-L1 expression in the 4T1 cell model, and tri-therapies with the further addition of 5-Aza or entinostat fully eradicated the tumors[49,50]. Indeed, the authors showed that the use of epigenetic drugs led to the elimination of the potent immunosuppressive myeloid-derived suppressor cells (MDSCs) at very low concentrations, doses generally well tolerated by cancer cells. Moreover, the addition of antibodies targeting MDSCs, or the use of a PI3K inhibitor known to reduce circulating MDSCs, in combination with immunotherapy led to the same efficiency. Similar results were observed in NSCLC cells treated with Mocetinostat (one inhibitor of HDAC I/IV classes)[51]. On the opposite, HDAC11-/- knock-out mice presented very high levels of immunosuppressive MDSCs suggesting that targeting only one epigenetic enzyme may lead to opposite results although molecular mechanisms remain unknown.
Many previous studies demonstrated the central role of epigenetic modifications on CD4+ T-cell polarization and therefore on cancer prognosis and outcome. Indeed, while Th1 polarization is associated with a good prognosis, Tregs (CD4+, FOXP3+) are immunoregulators whose presence in the tumor immune infiltrate is associated with a repression of CD8+ T-cells and NKs activity via the production of IL-10, IL-35 and TGFβ. Epigenetic drugs might also activate FOXP3 expression[52] and immunotolerance and thus might limit the benefits of immunotherapy. However, it has been recently reported, in the breast cancer 4T1 and in NSCLC cells, that the use of large spectra HDACi or Mocetinostat increased PD-L1 expression and conconmitantly down-regulate T-regs leading to increased CD8+ T-cell infiltration and survival in mice suggesting that Tregs might also be inhibited by epidrugs[50,51].
Cytosolic NADP+-dependent isocitrate dehydrogenase 1 and glioma
LGGs associated with a wild type cytosolic NADP+-dependent isocitrate dehydrogenase 1 (IDH1) gene, present a very aggressive phenotype which is comparable to the high grade glioma glioblastoma (GBM). Moreover, the tumors with a non-mutated IDH1 locus present a more immunosuppressive context, than those carrying a mutated IDH1 gene, and expressed higher levels of PD-L1 correlated to a lower level of PD-L1 promoter methylation than the one observed in the tumors with the mutated form of the gene[32,53,54]. These data could be explained by the fact that the mutated IDH1 protein produces higher levels of 2-hydroxyglutarate, a specific sub-product which favors methylation and repression of the PD-L1 promoter. Altogether, these data suggest that some specific subtypes of LGGs may respond differently to anti-checkpoint inhibitor therapies. We could then speculate that the use of anti-PD-L1 immunotherapies may improve conventional treatment outcomes when used in patients diagnosed with a LGG carrying a non-mutated IDH1 gene but not in tumors carrying the mutated version of the gene. For these last patients, these treatments should be combined with DNA demethylating agents. In addition, expression of PD-L1 mRNA in GBM has also been associated with non-mutated IDH1 gene and poor prognosis suggesting that overall survival of these patients could be significantly improved by anti-PD-L1 therapies[55].
Clinical trials
Interestingly, therapy resistance linked to demethylating agents which are used in several first line anti-cancer therapies and which induce the expression of PD-L1 could be annihilated in the second line of treatment by an anti-PD-L1 strategy. Phase I/II clinical trials including patients with different cancer origins (NSCLC, diffuse large B-cell lymphoma, metastatic or normal breast cancer, melanoma, squamous cell carcinoma of the oral cavity, colorectal cancer, HNSCC, renal carcinoma, urothelial carcinoma, lymphoma, leukemia, ovarian carcinoma, MDS) and treated with demethylating agents (5-Aza or 5-AzaCdR) or HDACi (vorinostat or entinostat) combined with ICB therapy are currently in progress and summarized in[56]. For example, the NCT02961101 phase1/2 trial is recruiting patients to analyze the effects of a treatment combining a low dose of decitabine with anti-PD-1 in relapsed or refractory malignancies in cancer of different origins.
Promising new combinations of epidrugs with anti-PD-1/PD-L1
Numerous clinical trials showed a large resistance to anti-PD-1 treatment in TNBC and these data were confirmed in mouse models since anti-PD-1 antibodies also failed to produce an objective response in TNBC xenograph models[57]. However, invalidation of the lysine demethylase LSD1 which catalyzes H3K4 and H3K9 demethylation, using specific inhibitors such as HCI-2509 or tranylcypromine, combined with anti-PD-1 antibodies significantly decreased xenograph tumor growth and metastasis and increased the number of TILs. Another study reported that contrary to breast cancer, 60% to 90% of pancreatic carcinoma expressed PD-L1[58]. Interestingly, the authors showed in pancreatic cancer cell models that this expression was controlled by the HMT mixed lineage leukemia 1 (MML1) enzyme which catalyzes the trimethylation of histone H3 lysine 4 (H3K4me3), and that targeted inhibition of HMTs, using Verticillin A, enhanced the anti-PD-L1 response in these models.
Tumor antigen and future epidrugs/immunotherapies combinations
Most of tumor antigens are proteins normally expressed in the male germline lineage which are abnormally expressed in tumor cells. These tumor antigens, whose the most studied are melanoma-antigen family A and NY-ESO-1 (New York-esophageal squamous cell carcinoma-1), are presented by MHC-I on tumor cells and are targets and activators of immune cells. Interestingly, epigenetic modifications have been linked to the control of the expression of most of these antigens and demethylating agents or HDACi increased their expression in various models (for a review, see[59]). For example, in in vitro cancer cells models (glioma: U251 and mesothelioma: Meso96), repression of NY-ESO-1 gene expression was regulated by a complex mechanism involving different DNMTs and HDAC1-including complexes which were recruited sequentially[60]. Combination of anti-disialoganglioside (GD2), which targets the tumor antigen GD2, with vorinostat, one HDACi, also decreased neuroblastoma tumor growth in mice[61]. In these tumors, the HDACi treatment led to an increase in M2-like macrophage infiltration in the tumor but decreased immunosuppressive MDSCs.
Moreover, MHC gene expression is also epigenetically controlled. Indeed, hypermethylation of MHC promoters in melanoma was correlated with decreased antigen presentation. Immunoedition in cancers could directly contribute to immunotherapy resistance since this process drives the selection of stem cell-like immune cells associated with a high resistance to treatments. Indeed, the expression of the pluripotent transcriptional factor NANOG in these cells can bind and activate the HDAC1 promoter and, consequently, induce the deacetylation and repression of several target genes, such as NOXA, therefore leading to stem cell-like phenotype and treatment resistance[62]. Combining HDACi with immunotherapies in NANOG-positive tumors may therefore limit cancer relapse and become a new line of treatment.
Stimulator of interferon genes (STING), is a protein normally activated by the cyclic GMP-AMP synthase which detects exogenous cytosolic DNA following bacterial or viral cell infection or DNA release by tumor cells. Expression of the lysine demethylase KDM5B has been shown to be inversely correlated to STING, in ovarian cystadenocarcinoma, bladder urothelial carcinoma and breast invasive carcinoma and inversely correlated to CD8+ T-cell infiltration and survival, as well[63]. This recent study also revealed that KDM5B can demethylate histones and repress STING promoter and that this effect was specific since it was inhibited by KDM5Bi. This new set of data suggests that KDM5Bi could be tested in the future to restore T-cell infiltration and therefore improve anti-PD-1/PD-L1 treatments.
Transgenic TCR and epidrugs combination for future therapies
New protocols of engineering in immunology have recently allowed to develop novel cell-based immunotherapies. Adoptive transfer of TCR transgenic T-cells has become a promising approach in the field. For example, a protocol has been designed to target MART-1, which is a major antigen present in 95% of melanomas, and therefore to design engineered T-lymphocytes which produce a transgenic TCR specific of MART-1. However, it has been shown that these strategies had no effect on melanomas presenting a resistance to BRAF inhibitors. Indeed, these cells present important defects in the apoptotic machinery which impair TCR-induced apoptosis but it has also been shown that HDACi, such as SAHA, can regulate apoptotic gene expression and restore TCR-induced tumor cell death. Even if the mechanisms and pathways involved remain still unclear, preliminary results suggested that HDACi modulated the expression of apoptotic genes, restoring the susceptibility of melanoma cells to apoptosis.
Conclusion
Altogether, these recent data clearly demonstrate that, although immune checkpoints and particularly PD-L1 are frequently overexpressed in tumors compared to healthy tissues, some factors might still be able to modulate their expression via the regulation of DNA methylation and histone modifications of their promoters [Figure 2]. Since immunohistochemistry staining are still difficult to quantify and standardize, mainly because of different antibody specificities, various fixation times and partially subjective interpretations between different laboratories, we speculate that quantification of promoter methylation of immune checkpoint inhibitors could be a robust and reproducible alternative method to quickly select good responsive patients which could be directed towards anti-PD-L1 therapies and those who could be good candidates for combined therapy using DNA demethylating agents and immunotherapy protocols. But, further pre-clinical and clinical studies testing the combination of ICB with epidrugs and additional regulators of immune checkpoint expression will be necessary to evaluate their efficiency to bypass the immunotherapy resistance in cancers.
Declarations
AcknowledgementsPeixoto P, Boyer-Guittaut M and Hervouet E are supported by funding from institutional grants from INSERM, EFS and Univ. Bourgogne Franche-Comté and by the «Ligue Contre le Cancer» (2017), «Cancéropôle Grand-Est» (OPE-2017-0052/DOC) and «Région Bourgogne-Franche-Comté» (2018).
Authors’ contributionsWrote the paper: Peixoto P, Hervouet E
Edited the paper: Renaude E, Boyer-Guittaut M
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by funding from institutional grants from INSERM, EFS, and University of Bourgogne Franche-Comté and by the “Ligue Contre le Cancer” (2017) and the “Région Bourgogne Franche-Comté” (2014C-15449).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015;527:249-53.
2. Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Egeler RM, et al. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J Pathol 2011;223:347-57.
3. Hirano S, Iwashita Y, Sasaki A, Kai S, Ohta M, et al. Increased mRNA expression of chemokines in hepatocellular carcinoma with tumor-infiltrating lymphocytes. J Gastroenterol Hepatol 2007;22:690-6.
4. de la Cruz-Merino L, Barco-Sánchez A, Henao Carrasco F, Nogales Fernández E, Vallejo Benítez A, et al. New insights into the role of the immune microenvironment in breast carcinoma. Clin Dev Immunol 2013;2013:785317.
7. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293-7.
8. Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Mol Oncol 2014;8:1132-9.
9. Kovacs D, Migliano E, Muscardin L, Silipo V, Catricalà C, et al. The role of Wnt/β-catenin signaling pathway in melanoma epithelial-to-mesenchymal-like switching: evidences from patients-derived cell lines. Oncotarget 2016;7:43295-314.
10. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017;46:197-204.
11. Collins DC, Chenard-Poirier M, Lopez JS. The PI3K pathway at the crossroads of cancer and the immune system: strategies for next generation immunotherapy combinations. Curr Cancer Drug Targets 2018;18:355-64.
12. Lu C, Talukder A, Savage NM, Singh N, Liu K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017;6:e1291106.
13. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011;117:4501-10.
14. Yoneda K, Imanishi N, Ichiki Y, Tanaka F. Immune checkpoint inhibitors (ICIs) in non-small cell lung cancer (NSCLC). J UOEH 2018;40:173-89.
15. Schachter J, Ribas A, Long GV, Arance A, Grob JJ, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017;390:1853-62.
16. Chatwal MS, Tanvetyanon T. Malignant mesothelioma clinical trial combines immunotherapy drugs. Immunotherapy 2018;10:341-4.
17. Massari F, Santoni M, Ciccarese C, Santini D, Alfieri S, et al. PD-1 blockade therapy in renal cell carcinoma: current studies and future promises. Cancer Treat Rev 2015;41:114-21.
18. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014;515:558-62.
19. Rebelatto MC, Midha A, Mistry A, Sabalos C, Schechter N, et al. Development of a programmed cell death ligand-1 immunohistochemical assay validated for analysis of non-small cell lung cancer and head and neck squamous cell carcinoma. Diagn Pathol 2016;11:95.
20. Meyers DE, Bryan PM, Banerji S, Morris DG. Targeting the PD-1/PD-L1 axis for the treatment of non-small-cell lung cancer. Curr Oncol 2018;25:e324-34.
21. Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol 2016;34:2460-7.
22. Hervouet E, Cheray M, Vallette FM, Cartron PF. DNA methylation and apoptosis resistance in cancer cells. Cells 2013;2:545-73.
23. Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
24. Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 2013;4:2067-79.
25. Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011;1:598-607.
26. Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol 2011;29:1949-55.
27. Goode EL, Block MS, Kalli KR, Vierkant RA, et al; Ovarian Tumor Tissue Analysis (OTTA) Consortium. Dose-response association of CD8+ tumor-infiltrating lymphocytes and survival time in high-grade serous ovarian cancer. JAMA Oncol 2017;3:e173290.
28. Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One 2013;8:e62924.
29. Sasidharan Nair V, El Salhat H, Taha RZ, John A, Ali BR, et al. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin Epigenetics 2018;10:78.
30. Bensaid D, Blondy T, Deshayes S, Dehame V, Bertrand P, et al. Assessment of new HDAC inhibitors for immunotherapy of malignant pleural mesothelioma. Clin Epigenetics 2018;10:79.
31. Sasidharan Nair V, Toor SM, Taha RZ, Shaath H, Elkord E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin Epigenetics 2018;10:104.
32. Röver LK, Gevensleben H, Dietrich J, Bootz F, Landsberg J, et al. PD-1 (PDCD1) promoter methylation is a prognostic factor in patients with diffuse lower-grade gliomas harboring isocitrate dehydrogenase (IDH) mutations. EBioMedicine 2018;28:97-104.
33. Marwitz S, Scheufele S, Perner S, Reck M, Ammerpohl O, et al. Epigenetic modifications of the immune-checkpoint genes CTLA4 and PDCD1 in non-small cell lung cancer results in increased expression. Clin Epigenetics 2017;9:51.
34. Zhang Y, Xiang C, Wang Y, Duan Y, Liu C, et al. PD-L1 promoter methylation mediates the resistance response to anti-PD-1 therapy in NSCLC patients with EGFR-TKI resistance. Oncotarget 2017;8:101535-44.
35. Goltz D, Gevensleben H, Vogt TJ, Dietrich J, Golletz C, et al. CTLA4 methylation predicts response to anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients. JCI Insight 2018; doi: 10.1172/jci.insight.96793.
36. Goltz D, Gevensleben H, Dietrich J, Ellinger J, Landsberg J, et al. Promoter methylation of the immune checkpoint receptor PD-1 (PDCD1) is an independent prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients following radical prostatectomy. Oncoimmunology 2016;5:e1221555.
37. Goltz D, Gevensleben H, Grünen S, Dietrich J, Kristiansen G, et al. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia 2017;31:738-43.
38. Wu J, Xu X, Lee EJ, Shull AY, Pei L, et al. Phenotypic alteration of CD8+ T cells in chronic lymphocytic leukemia is associated with epigenetic reprogramming. Oncotarget 2016;7:40558-70.
39. Asgarova A, Asgarov K, Godet Y, Peixoto P, Nadaradjane A, et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018;7:e1423170.
41. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, et al. De novo epigenetic programs inhibit pd-1 blockade-mediated t cell rejuvenation. Cell 2017;170:142-57.
42. Ørskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, et al. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: a rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015;6:9612-26.
43. Duruisseaux M, Martínez-Cardús A, Calleja-Cervantes ME, Moran S, Castro de Moura M, et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir Med 2018;6:771-81.
44. Rieke DT, Ochsenreither S, Klinghammer K, Seiwert TY, Klauschen F, et al. Methylation of RAD51B, XRCC3 and other homologous recombination genes is associated with expression of immune checkpoints and an inflammatory signature in squamous cell carcinoma of the head and neck, lung and cervix. Oncotarget 2016;7:75379-93.
45. Franzen A, Vogt TJ, Müller T, Dietrich J, Schröck A, et al. PD-L1 (CD274) and PD-L2 (PDCD1LG2) promoter methylation is associated with HPV infection and transcriptional repression in head and neck squamous cell carcinomas. Oncotarget 2018;9:641-50.
46. Goltz D, Gevensleben H, Dietrich J, Schroeck F, de Vos L, et al. PDCD1 (PD-1) promoter methylation predicts outcome in head and neck squamous cell carcinoma patients. Oncotarget 2017;8:41011-20.
47. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014;28:1280-8.
48. Lai Q, Wang H, Li A, Xu Y, Tang L, et al. Decitibine improve the efficiency of anti-PD-1 therapy via activating the response to IFN/PD-L1 signal of lung cancer cells. Oncogene 2018;37:2302-12.
49. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A 2014;111:11774-9.
50. Terranova-Barberio M, Thomas S, Ali N, Pawlowska N, Park J, et al. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017;8:114156-72.
51. Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother 2018;67:381-92.
52. Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med 2007;204:1543-51.
53. Mu L, Long Y, Yang C, Jin L, Tao H, et al. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front Mol Neurosci 2018;11:82.
54. Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol 2017;19:1460-8.
55. Lee KS, Lee K, Yun S, Moon S, Park Y, et al. Prognostic relevance of programmed cell death ligand 1 expression in glioblastoma. J Neurooncol 2018;136:453-61.
56. Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics 2017;9:59.
57. Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2018; doi: 10.1038/s41388-018-0451-5.
58. Lu C, Paschall AV, Shi H, Savage N, Waller JL, et al. The MLL1-H3K4me3 Axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst 2017; doi: 10.1093/jnci/djw283.
59. Arenas-Ramirez N, Sahin D, Boyman O. Epigenetic mechanisms of tumor resistance to immunotherapy. Cell Mol Life Sci 2018;75:4163-76.
60. Cartron PF, Blanquart C, Hervouet E, Gregoire M, Vallette FM. HDAC1-mSin3a-NCOR1, Dnmt3b-HDAC1-Egr1 and Dnmt1-PCNA-UHRF1-G9a regulate the NY-ESO1 gene expression. Mol Oncol 2013;7:452-63.
61. Kroesen M, Büll C, Gielen PR, Brok IC, Armandari I, et al. Anti-GD2 mAb and vorinostat synergize in the treatment of neuroblastoma. Oncoimmunology 2016;5:e1164919.
62. Song KH, Choi CH, Lee HJ, Oh SJ, Woo SR, et al. HDAC1 upregulation by NANOG promotes multidrug resistance and a stem-like phenotype in immune edited tumor cells. Cancer Res 2017;77:5039-53.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Peixoto P, Renaude E, Boyer-Guittaut M, Hervouet E. Epigenetics, a key player of immunotherapy resistance. Cancer Drug Resist 2018;1:219-29. http://dx.doi.org/10.20517/cdr.2018.17
AMA Style
Peixoto P, Renaude E, Boyer-Guittaut M, Hervouet E. Epigenetics, a key player of immunotherapy resistance. Cancer Drug Resistance. 2018; 1(4): 219-29. http://dx.doi.org/10.20517/cdr.2018.17
Chicago/Turabian Style
Peixoto, Paul, Elodie Renaude, Michaël Boyer-Guittaut, Eric Hervouet. 2018. "Epigenetics, a key player of immunotherapy resistance" Cancer Drug Resistance. 1, no.4: 219-29. http://dx.doi.org/10.20517/cdr.2018.17
ACS Style
Peixoto, P.; Renaude E.; Boyer-Guittaut M.; Hervouet E. Epigenetics, a key player of immunotherapy resistance. Cancer Drug Resist. 2018, 1, 219-29. http://dx.doi.org/10.20517/cdr.2018.17
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 12 clicks
Cite This Article 12 clicks

 Like This Article 0
likes
Like This Article 0
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.