
Drug targets and resistance mechanisms in multiple myeloma


Abstract
Multiple myeloma (MM), a malignancy of plasma cells, is the second most prevalent blood cancer (10%). A PubMed search has been conducted for English research papers and reviews published until January 2018. Numerous drugs are used in treatment of MM. These include the antineoplastic alkylating agents cyclophosphamide, busulfan and melphalan, immunomodulators such as lenalidomide and thalidomide, corticosteroids including dexamethasone, microtubule-targeting agents, such as paclitaxel and vinca alkaloids, as well as the proteasome inhibitors bortezomib and carfilzomib. Despite the considerable number of treatment options, MM is still difficult to treat, which is mirrored by the poor 10-year survival rate of 3%. Resistance to chemotherapy is often the cause for therapy failure. These resistances can be due to the overexpression of efflux pumps, genetic and epigenetic aberrations and the microenvironment of MM. With the gain of knowledge regarding genetic and molecular changes, many molecular targeted therapies including cell signaling targeted therapies are being developed against relapsed/refractory MM. Additionally, epigenetic aberrations such as DNA methylation and histone modifications steered MM management in new directions. Amongst these novel targeted therapies, inhibitors of histone deacetylase, Aurora kinase, inhibitors of the PI3K/AKT/mTOR pathway and cyclin dependent kinases are promising.
Keywords
Introduction
Multiple myeloma (MM) is the second most prevalent hematological malignancy worldwide, with a median onset of 60 years. This incurable malignancy develops from an accumulation of terminally differentiated monoclonal plasma cells (PC) in the bone marrow. In the United States, the lifetime risk for MM is one in 143 (0.7%)[1]. Additionally, MM is associated with several severe impairments such as lytic bone lesions, osteoporosis, compression fractures, bone pain and patient immobility. Due to the amount of malignant monoclonal plasma cells, the immune system and the blood formation of patients are impaired[2]. The median overall survival of patients with newly diagnosed MM was 2.5-3 years[3], but could be extended with new drugs such as proteasome inhibitors (PIs) to 4-5 years. Nevertheless, the 10-year survival rate is only 3%[4,5]. A significant contributor to treatment failure leading to clinical relapse is the emergence of multi-drug resistance (MDR). MDR represents a phenomenon, whereby the cancer cells are resistant to a wide variety of structurally and functionally unrelated drugs following exposure to a single chemotherapeutic agent. Cancers can either are inherently untreatable or become resistant to a wide variety of anticancer drugs and their combinations[6]. Most MM patients respond successfully to initial induction therapy. However, all patients eventually relapse. Genetic research revealed that MM is a heterogeneous disease complicating treatment and demonstrating the need for individualized therapy options. Here, we provide an overview of the different classes of targets in MM and the factors that influence search for new drugs. We also discuss the influence of MDR on treatment of MM.
A PubMed search for “multiple myeloma” and “cancer” and “multidrug resistance” has been conducted for literature published until January 2018. The search was limited to English language documents where full text was available.
Treatment targets in MM
Targeting the proteasome
The ubiquitin-proteasome (UPS) pathway is the central non-lysosomal pathway for protein degradation in eukaryotic cells. The proteasome is a large, multi-subunit complex that is located in the cytosol and the nucleus. The proteasome controls degradation of intracellular proteins by the attachment of a poly-ubiquitin chain, followed by a rapid degradation into small peptides[7]. This process is essential for protein homeostasis. It affects key cellular processes such as intracellular protein processing and degradation, regulation of cell cycle progression, apoptosis, antigen presentation, and transcription[8]. The constitutive 26S proteasome consists of two outer 19S regulatory particles and an inner 20S core particle with two identical rings of seven α-subunits and two identical rings of seven β-subunits. The β-subunits recognize and unfold the ubiquitin-bound proteins for entry into the proteasome. The catalytically active subunits β1, β2, and β5 catalyze proteolysis. The remnants of protein degradation can either be used for presentation on major histocompatibility complex (MHC) class I molecules on the cell surface to initiate an immune response or be recycled for protein synthesis[9,10]. The disruption of proteasome activity leads to rapid accumulation of toxic regulatory proteins that activate the unfolded protein response (UPR), thereby blocking protein translation and initiating alternative degradation pathways. If the cellular stress cannot be overcome by UPR, cells commit apoptosis[11]. Another kind of proteasome is the immunoproteasome. The immunoproteasome is a unique target found in lymphoid-derived cells. The use of specific immunoproteasome inhibitors could preserve efficacy while reducing toxicities[12]. But until now, no specific inhibitors are on the market[13].
The activity of the proteasome is upregulated in MM resulting in excessive degradation of specific substrates of relevance, including the tumor suppressor p53 and the inhibitor of nuclear factor-κB (NF-κB), IκB. Tumor progression is driven by oncogenic transformation, the upregulation of pro-inflammatory cytokine signaling via tumor necrosis factor-alpha (TNFα) and interleukin-6 (IL-6). Cell adhesion is favored through the ICAM-1 and VCAM-1 molecules. Other pathways included are anti-apoptotic signaling via Bcl-2, IAP, and TRAIL and proangiogenic signaling via VEGF and GRO-α. MM cells are rapidly proliferating. PIs target one or more subunits of the 20S proteasome. These agents have different chemical warheads and different inhibitory properties. Bortezomib (BTZ) was the first PI, approved in 2008, with a peptide boronic acid warhead. It is a slowly reversible inhibitor of the β5 catalytic subunit. BTZ is approved for administration via intravenous bolus or subcutaneous injection. Next generation PIs were developed to enhance the bioavailability, achieve reduced toxicity and to overcome resistance. Carfilzomib (CFZ) is a peptide epoxyketone that irreversibly inhibits the β5 site[14]. The third approved proteasome inhibitor, ixazomib, is also the first orally administered agent. Ixazomib prolonged progression-free survival in a study comparing ixazomib-lenalidomide-dexamethasone (IRd) with placebo-lenalidomide-dexamethasone[15]. Another study proved that oncology patients prefer oral treatment compared to the intravenous route, suggesting another advantage for ixazomib[16]. A new oral proteasome inhibitor is oprozomib, which is currently under investigation[17]. Likewise carfilzomib, it is an irreversible peptide epoxyketone. The investigational agent marizomib, a marine natural product, acts via irreversible inhibition of multiple catalytic sites within the 20S core[18-20]. The clinical development of proteasome inhibitors is increasing. The treatment with PIs significantly prolonged patient lifetime[21].
Targeting signal transduction pathways
PI3K/AKT/mTOR signaling pathway
The PI3K/AKT/mTOR pathway is an intracellular signaling route regulating the cell cycle. Phosphatidylinositol 3-kinase (PI3K), AKT (a serine/threonine kinase also known as PKB), and mammalian target of rapamycin (mTOR) are three major junctions in this pathway and are typically activated by upstream signaling of tyrosine kinases and other receptor molecules such as hormones and other mitogenic factors. It is a gatekeeper for tumor growth. It is activated because of loss of function of tumor suppressor genes. Moreover, the PI3K/AKT/mTOR pathway mediates cytokine-induced MM cell proliferation, survival and development of drug resistance[22,23]. The activation of PI3K leads to the recruitment of serine-threonine kinase AKT to the cell membrane to catalyze their function. This enables the phosphorylation of multiple downstream targets including mTOR, which in turn regulates protein synthesis and translation[24]. Many studies focus on mTOR inhibitors and their role in MM treatment. mTOR is a serine-threonine protein kinase that belongs to the PI3K-related kinase family. It plays a central role in regulating critical cellular processes such as growth, proliferation, cytoskeletal organization, transcription, protein synthesis and ribosomal biogenesis[25]. mTOR exists as two separate complexes, TORC1 and TORC2. The most established mTOR inhibitors are rapalogues such as rapamycin. They primarily inhibit TORC1, but TORC2 is actually a key activator of AKT. This started the quest to find inhibitors that inhibit both complexes[26]. CCI-779 (temsirolimus) is an analogue of rapamycin, which showed anti-MM activity in vitro and in vivo. CCI-779 showed efficacy in a clinical phase II study of MM and demonstrated synergy in combination with lenalidomide in vitro. A phase I trial of lenalidomide in combination with CCI-779 in patients with relapsed MM revealed more adverse events than expected, and the pharmacokinetic analysis suggested a drug-drug interaction[27]. In 2014, Yee et al.[28] published data of a phase I clinical trial with lenalidomide and everolimus in relapsed or refractory MM. The combination of both drugs was well tolerated and showed responses in a heavily pretreated population.
AKT plays an integral role in MM pathogenesis, progression and resistance to standard treatments. Many upstream signaling pathways converge on AKT to mediate pro-survival signaling and in turn AKT suppresses apoptosis. Many AKT inhibitors are in preclinical and clinical development. Afuresertib, an oral AKT inhibitor showed a favorable safety profile and demonstrated clinical activity against MM[29]. A phase I study tested a combination of the MEK inhibitor trametinib with the AKT inhibitor afuresertib in patients with solid tumors and MM. However, clinically meaningful doses could not be achieved[30]. A phase II study of trametinib and the AKT inhibitor GSK2141795 in relapsed or refractory MM evaluated the antitumor activity of trametinib determined by overall response rate (ORR)[31]. A phase II trial investigated the AKT inhibitor AZD5363 in a targeted therapy directed by genetic testing in MM patients[32]. ONC201 is an orally bioavailable first-in-class small molecule. There is an ongoing phase I/II open-label study of ONC201, administered orally once every week, in combination with dexamethasone to patients with relapsed/refractory MM. The end of the study is estimated for March 2020[33]. A multi-center phase I/II clinical trial of the oral AKT inhibitor perifosine in combination with BTZ ± dexamethasone has demonstrated encouraging activity in heavily pretreated BTZ-exposed relapsed/refractory MM patients[34].
Acquired resistance to PI3K-mTOR inhibition was observed in patients with advanced renal cell cancer. Resistant cells over-expressed mTORC1 component RAPTOR at the mRNA and protein level. RAPTOR up-regulation contributes to PI3K-mTOR inhibitor resistance, and suggests that RAPTOR expression should be included in the pharmacodynamic assessment of mTOR kinase inhibitor trials[35].
Ras/Raf/MEK/Erk signaling pathway
The Ras/Raf/MEK/extracellular signal regulated kinase (Erk) signaling pathway is an important mediator for cellular survival, proliferation, angiogenesis and migration. Ras protein subfamilies play a key role for cellular signal transduction. Approximately 30% of malignancies involve Ras proto-oncogene mutations. Ras mutations may be present in 23%-54% at diagnosis, but they increase to 45%-81% in relapsed or refractory cases[36]. The Ras mutation is associated with a more aggressive phenotype and shorter survival. The Ras/MAPK signaling pathway is characterized by the mammalian MAPK signal transduction networks consisting of the Ras protein, the Raf kinase (Map3K), the MAP2K kinases (MEK1 and MEK2) and pathway distil kinases Erk1 and Erk2. Genetic changes are also associated with MM. For example, the t (4; 14) translocation results in overexpression of fibroblast growth factor receptor 3, and further stimulates the Ras/MAPK pathway[37,38]. Numerous efforts have been made to target this pathway. Sorafenib, an orally available compound that predominantly acts by inhibition of Raf kinase and VEGF receptor 2 inhibits MM proliferation. It also upregulates AKT phosphorylation. In vitro studies showed synergistic effects of sorafenib in combination with rapamycin to inhibit mTOR[39,40]. A phase II trial assessed the activity and tolerability of sorafenib in patients with refractory or relapsed MM. All patients discontinued the treatment protocol. The overall survival at 12 months was 50% and median progression-free survival was 1.2 months. Further research should focus on combination therapies of sorafenib with standard treatments[41]. A phase I study was undertaken to define the toxicity and the maximum tolerated doses (MTD) of the combination of sorafenib and BTZ in patients with advanced solid tumors. The combination showed preliminary signs of efficacy, supporting phase II studies[42]. AZD6244 (selumetinib) is a potent MEK inhibitor that has demonstrated high activity in both in vitro and in vivo tumor models, and it was found to target both MM cells and osteoclasts in the bone marrow-derived macrophages (BMM)[43,44]. However, a phase II study used to determine the response rate of AZD6244 in patients with relapsed or refractory MM found that AZD6244 alone was tolerable and showed minimal activity[45]. It was suggested that AZD6244 can re-sensitize cells with acquired resistance to lenalidomide and pomalidomide in combination with dexamethasone, thus further suggesting that combination therapies with MEK inhibitors could enhance the efficacy of current MM therapies[44].
JAK/STAT signaling pathway
The Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway was originally discovered in the context of α-interferon (IFN-α), γ-interferon (IFN-γ), and interleukin 6 (IL-6) mediated downstream signaling. Activated JAK regulates cell proliferation, differentiation, migration, and apoptosis[46]. One explanation could be the silencing of the JAK/STAT suppressor genes, through the hypermethylation of SHP-1 and SOCS-1. An in vitro experiment correlated the repressed expression of SHP-1 and SHP-2 with a better response to the induction therapy[47]. Several approaches have been proposed to target this signaling pathway. For instance, the JAK2 inhibitor TG101209 induced dose- and time-dependent cytotoxicity in a variety of MM cell lines by inhibiting cell cycle progression and inducing apoptosis in MM cell lines and patients' derived plasma cells[48]. Ruxolitinib, a tyrosine kinase inhibitor, TM-233, a novel analogue of 1'-acetoxychavicol acetate, and cantharidin induced cell death in in vitro models of MM[49-51]. The gold compound auranofin induced apoptosis of human MM cells through both down-regulation of STAT3 and inhibition of NF-κB activity[52]. R115777, a non-peptidomimetic farnesyl transferase inhibitor, induced apoptosis and the phosphorylation of both STAT3 and ERK1/2 was blocked[53]. INCB16562 was developed as a novel, selective, and orally bioavailable small-molecule inhibitor of JAK1 and JAK2 decidedly selective over JAK3. Treatment of MM cells with INCB16562 potently inhibited IL-6-induced phosphorylation of STAT3[54]. Lycorine, a natural alkaloid found in different Amaryllidaceae, like the cultivated bush lily (Clivia miniata), inhibited cellular viability and induced cell death in MM cell lines. It arrested the cell cycle in the G0/G1 phase and inhibited JAK2/STAT signaling through upregulation of suppressor of cytokine signaling 1 (SOCS1). SOCS1 encodes a JAK-binding protein that regulates the JAK/STAT signal transduction pathway and is a key negative regulatory factor in MM, thus attenuating cytokine-initiated signal transduction[55]. IL-6, which was previously mentioned as important factor acting in MM in the PI3K/AKT/mTOR and Ras/Raf/MEK/Erk signalling pathways, also acts as a MM growth factor by stimulating IL-6 receptor (IL-6R) and triggers phosphorylation of STAT3 via JAK1. Inhibition of the IL-6R/STAT3 pathway induced apoptosis of certain MM cell lines. For instance, the pan-JAK inhibitor, tetracyclic pyridine 6 (P6) induced growth arrest and subsequent apoptosis of IL-6-dependent MM-derived cell lines[56]. Atiprimod, a cationic amphiphilic compound with anti-inflammatory activity, inhibited proliferation of MM cell lines by inhibition of STAT3[57]. An open-label study of the safety and efficacy of atiprimod treatment for patients with refractory or relapsed MM was completed, but no further results have been reported[58]. Attempts to target IL-6 in clinical studies were not as successful as expected[59]. A clinical trial with the anti-IL-6 monoclonal antibody siltuximab did not show any improvement in the complete response rate or long-term outcome of MM patients[60,61]. One reason could be that binding of MM cells to bone marrow stromal cells (BMSCs) triggers survival even after inhibition of the IL-6/gp130/STAT3 pathway, suggesting that other growth mechanisms are involved. Cyclic adenosine monophosphate (cAMP) signaling induces the death of MM cells by down-regulating Mcl-1 via the JAK/STAT pathway. Remarkably, the addition of exogenous IL-6 did not prevent the inhibition of JAK1/STAT3 or MM cell death induced by cAMP[62]. 3-Formylchromone inhibited STAT3 phosphorylation and nuclear translocation in vitro[63]. The orally bioavailable JAK1/2 inhibitor CYT387 prevented IL-6-induced phosphorylation of STAT3 and greatly decreased IL-6- and insulin-like growth factor-1-induced phosphorylation of AKT and extracellular signal-regulated kinase in vitro[64]. Serenoa repens, a palm species, down-regulated phosphorylated STAT 3 and IL-6 induced phosphorylated STAT 3 and extracellular signal-related kinase (ERK) in vitro[65]. In another in vitro study, the chalcone butein suppressed constitutive and inducible STAT3 activation and STAT3-regulated gene products through the induction of the protein tyrosine phosphatase SHP-1[66]. Nifuroxazide inhibits the constitutive phosphorylation of STAT3 in MM cells by reducing JAK kinase autophosphorylation and down-regulated the STAT3 target gene MCL1[67]. Tyrphostin/AG490 suppressed cell proliferation and induced apoptosis in IL-6-dependent MM cell lines. JAK2 kinase activity, ERK2 and STAT3 phosphorylation were inhibited[68].
Wnt/β-catenin signaling pathway
The canonical Wnt signaling pathway relies on the stabilization of cytosolic β-catenin. In the absence of Wnt protein, β-catenin is phosphorylated by glycogen synthase kinase3b (GSK-3b) and casein kinase 1 and targeted for ubiquitination and degradation by the proteasome. The binding of Wnt to its receptor inhibits the activity of GSK-3b, resulting in the accumulation of β-catenin in the cytoplasm. If the concentration of β-catenin has reached a certain level, it translocates to the nucleus, where it regulates the expression of canonical Wnt target genes[69]. The canonical Wnt/β-catenin signaling pathway participates in the pathogenesis of MM by regulating the differentiation, proliferation, apoptosis, and migration of MM cells[70]. β-Catenin is overexpressed in MM cells. The administration of recombined human programmed cell death 5 (rhPDCD5) promotes MM apoptosis induced by dexamethasone. The combination treatment of rhPDCD5 plus dexamethasone down-regulated the mRNA and protein expressions of Wnt effectors including β-catenin, TCF4, survivin and c-Myc[71]. MM cells promote the development of osteolytic bone lesions by producing Wnt signaling inhibitors such as Dickkopf-1 (DKK1) to suppress the osteoblast differentiation[72,73]. A single-arm, open-label, phase II clinical trial was conducted to evaluate the effect of BHQ880, anti-DKK1 neutralizing antibody in previously untreated patients with high-risk, smoldering MM (NCT01302886)[74]. Targeting Wnt signalling inhibits bone destruction and reduces tumour burden[75,76].
Dysregulation of Wnt/β-catenin pathway has been observed in lenalidomide-resistant cells. β-catenin accumulation and its translocation to nucleus caused increased cell survival. It is thus presumed that disruption of E3 ubiquitin ligase complex functionality after intensive thalidomide treatment causes β-catenin accumulation even when Wnt is bound, thus causing overexpression of various pro-survival and anti-apoptotic factors that may be responsible for the thalidomide/lenalidomide resistance[77]. Cereblon forms an E3 ubiquitin ligase complex with damaged DNA binding protein 1 (DDB1), Cullin-4A (CUL4A), and regulator of cullins 1 (ROC1). This complex ubiquitinates a number of other proteins. Ubiquitination results in reduced levels of fibroblast growth factor 8 (FGF8) and fibroblast growth factor 10 (FGF10). Thalidomide binds strongly to cereblon, thereby interfering with the E3 ubiquitin ligase functions. Studies showed that cereblon is essential for the anti-myeloma activity of immunomodulatory drugs and that dysregulation of Wnt/β-catenin pathway may be one of the possible reasons of lenalidomide resistance. The CTNNB1 (rs4533622) polymorphism was found to be associated with disease activity (stage of the disease) in patients with MM[78].
NF-kB signaling pathway
NF-κB is a transcription factor that is activated by the cytokines TNF-α, IL-1β, and CD40, as well as other chemokines and cell adhesion molecules. In MM cells, many genes that are targets of NF-κB are highly expressed. Until now, mutations in eleven genes involved in regulation of the NFkB pathway have been identified at least 17% of MM tumors. Translocations or amplification were found to increase transcription of five positive regulators, which include NIK and NFKB1 plus three TNFR (CD40, TACI, LTBR). In addition, there was one example of a translocation that deleted a negative regulatory region from NIK so that it was less susceptible to proteasomal degradation. The NF-κB signaling pathway is affected by both activating and inactivating mutations in genes, such as TNF-receptor-associated factor (TRAF 2/3) and NF-κB-inducing kinase, as regulators of the non-canonical NF-κB signaling pathway. These mutations have been identified in 20% of MM patients, resulting in activation of NF-κB signaling without ligands, and may contribute to the progression of the disease[79]. Agents that inhibit NF-κB could be used to enhance the effects of conventional chemotherapeutic agents. BTZ seems to inhibit NF-κB activation indirectly by blocking proteasome degradation of IκBα (nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor α) and decreasing NF-κB nuclear translocation. The combination of BTZ, thalidomide and dexamethasone in transplant eligible patients showed an overall survival of 73% after a median follow-up of 30 months[80]. Patients having MM tumors with a strong NF-kB signature or with low level of TRAF3 RNA showed better response to bortezomib[81]. Moreover, MM cells with low levels of TRAF3 showed resistance to dexamethasone treatment[82]. Treatment of cells with dexamethasone induces expression of IkBα, which inhibits the classical pathway, and cIAP, which might block activity of the alternative NF-kB pathway. However, abnormalities of TRAF2, TRAF3 or cIAP1/2, might minimize the effect of increased cIAP1/2 and decrease the effectiveness of dexamethasone. Hence, dexamethasone treatment for patients that have mutations in genes regulating the alternative NFkB pathway might not be favourable[79].
RANK/RANKL/OPG signaling pathway
Osteolytic bone disease in MM results from increased osteoclastic activity, which is the consequence of overproduction of cytokines and chemokines that regulate osteoclast differentiation and activity. Receptor activator of NF-κB ligand (RANKL), its receptor RANK, and the endogenous soluble RANKL inhibitor, osteoprotegerin (OPG) regulate formation, function and survival of osteoclasts[83]. The interaction between RANKL and RANK/OPG plays a leading role in osteoclast activation and in plasma cell survival in MM[84]. Bone lesions and osteoporosis are common accompanying symptoms in MM. A phase III trial compared the human monoclonal antibody denosumab with zoledronic acid in patients with bone metastases and solid tumors or MM. Denosumab was superior to zoledronic acid for the prevention of skeletal-related events as primary end point[85]. A clinical phase I study tested the safety of AMGN-0007, a recombinant OPG construct, in MM and other cancer patients with bone lesions. A single subcutaneous dose of AMGN-0007 suppressed bone resorption in MM and breast carcinoma patients. Changes were comparable to those with pamidronate. AMGN-0007 was well tolerated[86]. The combination of intermediate doses of thalidomide with dexamethasone was effective in patients with refractory/relapsed MM and improved abnormal bone remodeling through the reduction of the sRANKL/OPG ratio. Soluble RANKL, OPG and osteopontin (OPN) were markers of bone resorption and formation and measured before and during treatment. Before treatment, patients had increased levels of sRANKL/OPG ratio, bone resorption markers and OPN, while they had suppressed bone formation. The administration of thalidomide and dexamethasone resulted in a significant reduction of sRANKL/OPG ratio, and bone resorption[87]. In another study, the stimulation of RANKL in RANK-expressing cells increased multidrug resistance protein 1 (MDR1), breast cancer resistance protein (BCRP), and lung resistance protein 1 (LRP1) expression and decreased Bim expression[88].
Targeting the cell cycle
Cell cycle defects are common features in human cancer cells. These defects include unscheduled proliferation, genomic instability (GIN) e.g., increased DNA mutations and chromosomal instability (CIN) such as changes in chromosome number. GIN is the result of alterations in the DNA damage response pathways, causing cell cycle progression in the presence of DNA damage. Aneuploidy and other chromosomal alterations (CIN) are caused by mutations in proteins involved in chromosome separation during mitosis. Proper chromosome separation is controlled by the SAC pathway, which shows frequently mutations in human cancer cells. Additional proteins have also been implicated in CIN, including Plk1 and aurora kinase A/B, Nek2, Cdc20, Cdc25, cyclins A/B/E and Cdk1.
Cdk
Cyclin D deregulation is a key hallmark in MM. The synthesis of cyclin D is initiated during G1 and drives the G1/S phase transition. Therefore, Cdks represent attractive therapeutic targets in MM. In general, inhibition of the interphase Cdks induces a cell cycle arrest or quiescence. On the other side, inhibition of Cdk1 results in a cell cycle arrest followed by either apoptosis or mitotic slippage. Cdk1 is essential for different processes in the cell cycle and it is likely that a strong inhibition of Cdk1 induces toxicity in normal cells. Therefore, the use of Cdk inhibitors is limited. Inhibition of other Cdks, such as the transcriptional Cdks Cdk7/9 prevents the phosphorylation of the carboxy-terminal domain of RNA polymerase II, resulting in decreased transcription of anti-apoptotic proteins and cell cycle regulators. This leads to a decrease in cellular levels of these proteins and subsequently induction of apoptosis[89].
Several pan-Cdk inhibitors have been investigated in MM[90]. First generation pan-Cdk inhibitors are flavopiridol and seliciclib[91]. Flavopiridol targets several kinases including Cdk1, Cdk2, Cdk4, Cdk6 and Cdk7. Seliciclib is slightly more specific by targeting Cdk2, Cdk7 and Cdk9. Both compete with ATP for the binding site on Cdks and blocking the formation of the activated kinase complex[92]. Flavopiridol and seliciclib induce apoptosis in MM cell lines. Studies with combination revealed that flavopiridol demonstrated synergistically enhancement with Bcl-2 antagonists, BTZ and TRAIL, while seliciclib was able to potentiate the anti-myeloma activity of doxorubicin and BTZ[93-96]. Another phase I/II trial tested the inhibitor palbociclib (PD-0332991) in sequential combination with BTZ and dexamethasone in relapsed/refractory multiple myeloma. BTZ was active at lower doses compared to other combination therapy studies and palbociclib was able to inhibit Cdk4/6 and the cell cycle initially in most patients[97]. Next generation pan-Cdk inhibitors are SNS-032, AT7519, dinaciclib, TG02, RGB-286638, LCQ195 and sangivamycin-like molecule 6 (SLM6). All these next generation pan-Cdk inhibitors inhibit both Cdk2 and Cdk9 and most of them inhibit Cdk1, Cdk5 and/or Cdk7. In addition, AT7519, RGB-286638 and LCQ195 also inhibit Cdk3, Cdk4 and/or Cdk6. Similar to flavopiridol and seliciclib, these pan-Cdk inhibitors induce cell death and reduce transcription efficiency in MM cells[91,98].
Microtubules
Microtubules are involved in the migration of the chromosomes during mitosis. Microtubules targeting agents (MTA) can be divided into two groups, the microtubule stabilizing agents that enhance the polymerization of microtubules and the destabilizing agents that inhibit polymerization of microtubules[99]. Inhibition with these drugs disrupts the normal microtubule dynamics and leads to an impaired formation of the spindle, chromosome alignment and SAC activation. They cause a cell cycle arrest, resulting either in cell death or in mitotic slippage[100].
The most tested and widely used MTA in MM therapy is vincristine[101]. However, the use of vincristine in MM patients is frequently associated with the development of multidrug resistance, and vincristine should therefore be replaced. Other MTAs such as paclitaxel and vinblastine or newer drugs such as docetaxel and vinorelbine showed little or no anti-myeloma activity[102,103]. Novel agents with microtubule-targeting activities are the isocourmarin derivate 185322[104], the 5HPP-33, CYT997 and PBOX-15 (pyrrol-1,5-benzoxazepine-15). Treatment with these agents results in M phase cell cycle arrest and induction of apoptosis in both established MM cell lines and primary cells[105]. The antihelmintic flubendazole inhibits microtubules by inhibited tubulin polymerization in vitro and in vivo by binding tubulin at a site distinct from vinblastine. Another advantage is that cells resistant to vinblastine because of overexpression of P-glycoprotein remained fully sensitive to flubendazole[106]. The substance KS99 inhibits the in vivo tumor growth of MM cells through the inhibition of BTK and tubulin[107]. STK405759 is another MTA with efficacy in resistant MM cells. The combination of STK405759 with BTZ, lenalidomide or dexamethasone demonstrated synergistic cytotoxic activity in mice[108].
Kinesin motor proteins
Kinesin motor proteins are key regulators of the mitotic spindle. For example, Eg5 is involved in both centrosome separation and bipolar spindle formation. Its inhibition results in monopolar spindles and mitotic arrest[109].
Filanesib (ARRY-520), a kinesin motor protein inhibitor showed in vivo synergy with pomalidomide plus dexamethasone treatment[110]. A phase I/II study of filanesib alone and in combination with low-dose dexamethasone in relapsed/refractory MM showed a manageable safety profile and encouraging activity in heavily pre-treated patients[111]. Another phase I dose-escalation study of filanesib plus bortezomib and dexamethasone in patients with recurrent/refractory MM indicated durable activity[112]. The Eg5 inhibitor SB743921 induces cell death also via inhibition of the NF-κB signalling. The combination with BTZ induced death in bortezomib-resistant KMS20 cells[113].
Aurora kinases
The family of Aurora kinases consists of three members, which are all involved in either mitosis (Aurora A and B kinase) or meiosis (Aurora C kinase). The inhibition of Aurora A and B kinase induces cell death[114]. Targeting Aurora A kinase induces mitotic spindle assembly defects, which result only in a transient arrest in mitosis. Aurora B kinase inhibition overrides the SAC causing polyploidy. Similarly to MTA, targeting aurora kinases result either in cell death or mitotic slippage causing tetraploid cells[115]. The Aurora A kinase inhibitor alisertib (MLN8237) is tested in a clinical trial phase I in combination with BTZ in patients with relapsed or refractory MM[116]. A phase II study tried to approve the efficacy of AT9283 in patients with relapsed or refractory MM[117].
p53
Activation of the tumor suppressor p53 results in cell cycle arrest and apoptosis. Therefore, therapeutic activation of p53 might be an attractive approach in MM[118]. Different mechanisms of p53 activation have been reported, such as reactivation by mouse double minute 2 homolog (MDM2) inhibition (e.g., Nutlin-3 and RITA) or restoring normal p53 function in p53-mutated cells (e.g., PRIMA-1 and MIRA-1)[119]. Until now, it is not possible to target TP53 deficiency with pharmacological agents. The attempt to activate another p53 family member, p73 with the drugs decitabine and melphalan did not work[120]. P53-related protein kinase (TP53RK) is an upstream kinase that mediates p53 activity. TP53RK knockdown inhibits p53 phosphorylation. The inhibition of TP53RK triggered MM cell apoptosis in vitro, suggesting that TP53RK could become a novel therapeutic[121].
Targeting histone deacetylases
Genes are epigenetically silenced by chromatin condensation. Histones are highly alkaline proteins that package and order DNA into structural units called nucleosomes. They are the chief protein components of chromatin, acting as spools around which DNA winds. Acetylation removes the positive charge on the histones, thereby decreasing the interaction of the N termini of histones with the negatively charged phosphate groups of DNA. Therefore, the condensed chromatin is transformed into a more relaxed structure that is associated with greater levels of gene transcription. Histone acetylation affects the DNA and chromatin structure, resulting in transcriptional activation of silenced genes. This activation is mediated by the enzymes histone acetyl transferase (HAT) and histone deacetylase (HDAC)[122]. In MM, the inhibition of histone deacetylation leads to DNA damage and upregulation of proteins that promote apoptosis and cell-cycle arrest. Inhibition of HDAC activity arrests the cell cycle and induces cell differentiation and cell death in MM cells. According to the four groups of histone proteins, HDACs are also divided into four classes. Inhibitors are available for class I and II HDACs. HDAC inhibition, by preserving or increasing histone acetylation, results in amplification of transcription factor activity[123].
Panobinostat is a nonselective HDAC inhibitor (HDACi) that inhibits class I, II, and IV HDACs at very low concentrations. It was approved as the first HDACi by the Food and Drug Administration (FDA) in 2015[124]. Vorinostat, an inhibitor of class I and II HDACs, induces MM cell death and reduces IL-6 production by bone marrow stromal cells. Vorinostat induces expression of p21 and p53 in MM cells, inhibits molecules with caspase inhibitory activity, inhibits proteasome activity, and induces cell death. In addition, vorinostat enhances the effectiveness of BTZ[125,126]. The precise mechanisms causing this synergy are not completely understood. One of the mechanisms is the dual inhibition of the proteasomal and aggresomal protein degradation pathways. BTZ inhibits proteasome and causes accumulation of proteins that form an aggresome by a process dependent on the interaction of HDAC6 with tubulin and dynein complex. HDAC6 inhibition leads to increased hyper-acetylation of tubulin and upregulation of polyubiquitinated proteins, resulting in apoptosis. A phase I study of CKD-581, a pan-histone deacetylase inhibitor, in patients with lymphoma or multiple myeloma refractory to standard therapy revealed that CKD-581 exhibited dose-proportional pharmacokinetics and modest anti-tumor efficacy[127].
Drug resistance mechanisms in MM
Resistance of cancer cells is a major obstacle in the treatment of MM. Multidrug- resistant cancer cells exhibit cross-resistance to a broad spectrum of structurally unrelated drugs. Drug resistance can be either intrinsic or acquired. Intrinsically drug-resistant cancer cells are insensitive to chemotherapy at the beginning of treatment. Tumor cells with acquired resistance are initially sensitive to particular chemotherapy regimens, but later they become non-responsive to the same or different anticancer drugs[128,129].
Role of ABC transporters for drug resistance
One of the reasons for drug resistance in MM patients is efflux transporters. P-glycoprotein (P-gp) is the best studied multidrug resistance protein. The multidrug resistance 1 (MDR1) gene encodes P-gp, which is the first known member of the ATP-binding cassette (ABC) transporter superfamily[130]. P-gp is extensively distributed and expressed in the intestinal epithelium, in liver cells, in the cells of the proximal tubule of the kidney, and in the capillary endothelial cells composing the blood-brain barrier. It acts as an efflux pump and provides cell protection against a plethora of xenobiotic and toxic substances. P-gp is a 170 kDa transmembrane glycoprotein[131]. Substrates enter P-gp either from the inner leaflet of the membrane or from an opening at the cytoplasmic side of the protein. The substrate is pumped out of the cell under consumption of ATP. ATP hydrolysis delivers the energy for extrusion of the substrate out of the cell[132,133]. In response to many chemotherapeutic substances, P-gp is up-regulated. This up-regulation causes a decrease in the intracellular accumulation of chemotherapeutic drugs, limiting their therapeutic efficacy. P-gp plays a role in resistance towards many drug classes such as proteasome inhibitors, anthracyclines, alkylating agents and immunomodulatory drugs[27,134-136]. Several studies demonstrated that MDR is not expressed in MM at diagnosis, but appears after treatment with these drugs[137,138]. P-gp is a predictive marker for therapy response and can be measured by whole body scans using imaging technologies[139,140]. Other members of transporter superfamily, which are important in drug resistance include multidrug resistance associated protein (MRP1), and breast cancer resistance protein (BCRP)[141,142]. Another trial suggested that other transporters such as ABCC4 (multidrug resistance-associated protein 4) and SLC29A2 may also hinder patients to achieve complete remission[143].
Single-nucleotide polymorphisms (SNPs) are associated with the development of malignant hematologic diseases. Different studies have been conducted to investigate correlations between distinct SNPs in ABC transporters and the risk for MM. The frequency of a distinct haplotype was significantly increased in Chinese MM patients compared with the control group[144]. A SNP variant in the ABCB1 gene was associated with a twofold higher risk for MM[145]. In contrast, another study did not find a correlation between P-gp activities or expression and MM subgroups[146]. Regarding BTZ-resistance, the putative role of the multidrug efflux transporter MDR1/P-glycoprotein expression has been investigated, however, its contribution to acquired BTZ-resistance remains unclear[147]. In contrast, acquired resistance to CFZ can be conferred by upregulation of Pgp[148].
Role of autophagy for drug resistance
Beside degradation via the proteasome, autophagy constitutes an alternative pathway for degradation and recycling of intracellular proteins. Autophagy functions by autophagosomes which sequester cytosolic proteins, followed by fusion with lysosomes for degradation. Autophagy is known to play a role in cancer[149] due to the deregulation of PI3K/Akt/mTOR signaling pathway and can mediate tumor cell survival under stress conditions[150]. A role in BTZ resistance was observed when autophagy-inducer activating transcription factor 4 (ATF4) was upregulated upon proteasome inhibition in different cancer cell lines[151]. Experiments, combining proteasome and autophagy inhibition showed synergistic cell death and higher levels of cell death than monotherapy[152]. Several autophagy targeting drugs have been tested, such as chloroquine in hepatocellular carcinoma and low-grade glioma, as well as fananserin, ticlopidine or valproic acid against cutaneous T cell lymphoma[153].
Role of MARCKS for drug resistance
The membrane protein myristoylated alanine-rich C-kinase substrate (MARCKS) is a protein that plays an important role in cell adhesion, spreading and invasion, and is thought to play an important role in metastasis. MARCKS is activated via phosphorylation by PKCs and modulates cell growth, cell-cycle progression and apoptosis. Association of PKC with drug resistance has been observed in different cancers including MM[154,155]. Nevertheless, diverse signaling effects of PKCs along with their large number of isoforms have made the development of drugs targeting these enzymes difficult. In an in vitro experiment, inhibition of MARCKS phosphorylation by enzastaurin significantly enhanced the sensitivity of resistant HMCLs and primary MM samples to bortezomib and to other anti-myeloma drugs[156].
Role of stem cells for drug resistance
MM is characterized by cancer stem cell-like cells (CSCs) that exhibit pronounced self-renewal and differentiation capacities, as well as pronounced drug resistance[157]. The MM CSCs receive both tumor cell-autonomous signals and signals from a variety of bystander cells in the tumor microenvironment (TME). Collectively, these signals trigger the self-renewal and survival of CSCs and the acquisition of drug resistance[158]. Until now, the existence of MM stem cells in multiple myeloma and the mechanisms of drug resistance are - despite a number of cited reports - disputed due to the lack of real markers for putative MM stem cells. Cellular markers significantly associated with a high rate of metastasis or recurrence such as side populations (SP) and ALDH1 positivity have been used to identify MM CSCs. Despite numerous attempts, the mechanisms of drug resistance remain unclear. Major pathways included are the Wingless (Wnt) pathway, the Hedgehog (Hh) pathway, Notch pathway, PI3K/Akt/mTOR and NF-κB pathway[70,159]. MM CSCs are further characterized by high drug efflux capacity, which is a common feature of stem cells. Moreover, the microRNA profile of MM CSCs is remarkably different from that of non-MM CSCs. Therefore, attempts are made to target MM CSCs microRNAs[160-162].
High expression of retinoic acid receptor α2 (RARα2) also resulted drug resistance[163]. The expression of anti-apoptotic Bcl-2 family members in MM CSCs increased RARα2 expression and drug resistance. Cellular quiescence is a property of hematopoietic stem cells and plays a crucial role in protecting stem cells[164]. Similar to normal stem cells, CSCs have slow cycling rates and are relatively quiescent. This property protects CSCs against chemotherapeutic agents that are most effective in dividing cells[165]. This property has been considered as a major mechanism of drug resistance.
Role of microenvironment
The microenvironment of MM is comprised of extracellular matrix components including collagens, laminin and fibronectin and cellular parts including bone marrow stem cells (BMSCs), osteoblasts, osteoclasts and others[166]. BMSCs secrete factors including IL-6, insulin-like growth factor 1 (IGF-1), RANKL, TNF-α, vascular endothelial growth factor (VEGF), and stromal cell-derived factor 1 α(SDF1)[167,168][Figure 1]. Crosstalk between canonical and non-canonical signaling pathways in MM cells and microenvironment may be responsible for the regulation of growth and survival of MM CSCs. Another form of de novo resistance is environment-mediated drug resistance (EMDR), in which the bone marrow microenvironment protects tumor cells from chemotherapy, radiotherapy or receptor-targeting drugs[169]. SDF-1 (CXCL12) is constitutively expressed and released by BMSCs and fibroblasts, while its receptor CXCR4 is expressed by MM cells. Activation of the SDF-1/CXCR4 axis promotes trans-endothelial migration, bone marrow homing, migration and adhesion of MM cells[170]. The MUC1 oncogene is known to confer tumor cells with resistance to apoptosis and necrosis. MUC1 expression was mediated by interleukin-6 and subsequent up-regulation of the JAK-STAT pathway[171]. MM cells acquire dormancy and drug resistance via interaction with bone marrow stroma cells (BMSC) in a hypoxic microenvironment. The CD180/MD-1 complex, a non-canonical LPS receptor, is expressed on MM cells but not on normal cells. It is markedly upregulated under adherent and hypoxic conditions and activates MM cells[172]. Bruton's tyrosine kinase (BTK) is a non-receptor tyrosine kinase that has been mainly studied in hematopoietic cells. BTK maintains self-renewal and drug-resistance of MM CSCs through AKT/WNT/β-catenin-dependent upregulation of key stemness genes. BTK high-expressing MM patients are associated with poor therapy outcome[173,174]. Cell adhesion mediated drug resistance (CAM-DR) is a mechanism, whereby MM cells escape the cytotoxic effects of anti-cancer therapy via adhesive interactions with BMSCs. Cancer cell binding to fibronectin adhesion molecules causes the CAM-DR phenotype. The HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A)/GG-PP (geranylgeranyl pyrophosphate)/Rho/Rho-kinase pathway mediates CAM-DR. The activation of Rho signaling in MM is the main stimulator of the Wnt3 pathway, which stimulates the tumor cells and is responsible for CAM-DR. Thus, the Wnt3/Rho signal causes the cells to enter a primed state phase in which the cells bind to stromal cells through the β1-integrin and eventually causes drug resistance[175]. Amongst others, it is achieved through adhesion molecules of the integrin family and CD138 (syndecan-1)[176]. Exosomes, which are responsible for the systemic cell-to-cell communication and regulate cell behavior by transferring mRNA, miRNAs and proteins, seem to play a major role in MM. Exosomes from BM mesenchymal stromal cells of MM patients facilitate MM progression and spreading, whereas exosomes from healthy people inhibit MM cell growth. Additionally, cancer patients possess higher amounts of exosomes, suggesting a future role as a biomarker[177,178]. Cancer associated fibroblasts (CAFs), modify the BM stroma and influence chemotaxis, adhesion, proliferation, and apoptosis of MM cells through cell-to-cell contact[179]. Insulin-like growth factor-1 (IGF-1) is a MM cell growth factor produced by plasma cells and the BM microenvironment. The abnormal overexpression of this cytokine and its receptor (IGF-1R) is associated with progression, severity, and unfavorable prognosis disease in MM patients. IGF-1 signaling is increased through IGF-1 secretion. IGF-1R activation is associated with BTZ resistance[180].
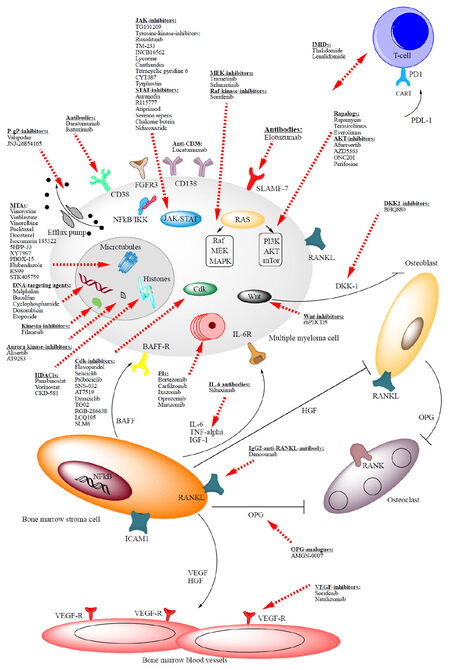
Figure 1. Overview of the MM cell interaction with its microenvironment and overview of MM drug targets. Red dashed arrows show targets. Bone-marrow stromal cells support growth and survival of MM cells via various cytokines. MM cells secrete dikkopf homolog 1 (DKK1) which inhibits the differentiation of osteoblastic precursors. Bone marrow stromal cells induce angiogenesis through secretion of VEGF. OPG produced by bone marrow stromal cells and osteblasts inhibits osteoclasts. BAFF-receptor: human B-cell-activating factor receptor; Cdk: cycline dependent kinase; DKK1: Dickkopf-1; HDACi: histone deacetylase inhibitor; HGF: hepatocyte growth factor; IL-6: interleukin-6; IL-6R: IL-6 receptor; JAK: Janus kinase; IMiD: immunomodulatory imide drug; MTA: microtubules targeting agents; mTOR: mammalian target of rapamycin; NF-κB: nuclear factor-κB; OPG: osteoprotegerin; PI: proteasome inhibitor; RANKL: receptor activator of NF-kappaB ligand; STAT: signal transducer and activator of transcription; TNFα: tumor necrosis factor-alpha; VEGF: vascular endothelial growth factor; WNT: wingless pathway
The inhibitor-of-apoptosis (IAP) family member, survivin, is one of the most significantly over-expressed genes in malignant cells. Survivin has been reported to inhibit apoptosis and regulate mitosis as well as cytokinesis. Studies indicate that survivin expression correlates with proliferation and poor overall survival of patients, as well as treatment failure[181,182]. XIAP (inhibitor of apoptosis protein) expression appears elevated in many cancer cells often conferring resistance to chemotherapy-induced cell death[183]. It was observed that maintenance of high IAP expression and low Smac/DIABLO expression after treatment lead to the doxorubicin-resistance of multiple myeloma cells[184]. Analysis of survivin and XIAP levels in response to chemotherapy with melphalan/prednisone or VMCP, showed an upregulation of IAP proteins and a postchemotherapeutic induction of MDR1 overexpression in more than half of the patients. Indicating that the overexpression of MDR proteins mediating the intracellular protein transport might facilitate the transport of IAPs and introduce the hyper-function of these proteins in these MM patients[185]. Myeloid cell leukemia-1 (Mcl-1) is a Bcl-2 family member. It enhances cell survival by inhibition of apoptosis. Mcl-1 expression was identified as underlying mechanism for myeloid-derived suppressor cell (MDSC) survival. MDSC are contributing to an immunosuppressive environment by their ability to inhibit T cell activity and thereby promoting cancer progression[186]. In addition, the vascular endothelial growth factor (VEGF) -induced MM cell proliferation and survival are mediated via Mcl-1[187].
Role of tumor heterogeneity for drug resistance
Clonal heterogeneity is a hallmark of solid tumors and hematological malignancies, and presents a significant obstacle for successful therapy. This complex genetic and phenotypic mosaic is determined to a large extent by accumulation of chromosome abnormalities (e.g., breaks, exchanges, duplications, etc.) and alterations in mutational load throughout the dynamic evolution of a tumor. In patients with acute myeloid leukemia, the presence of different cytogenetically defined subclones in the same patient has been associated with adverse outcome. Treatment with BTZ completely overcame the negative prognosis of high-risk cytogenetic aberrations in patients without subclones, but not in patients with additional subclonal cytogenetic aberrations[188]. So far, MM has been assumed to be a cancer entity characterized by dissemination of “fitter” clones replacing prior, less fit subclones. In this regard, spatial heterogeneity is a perplexing observation, challenging this current model. The non-homogeneous distribution of high-risk disease could result in misclassification of MM. This could be an explanation for the lack of sensitivity of currently used risk classifiers[189].
Role of genetics and genomics for drug resistance
Among the high-risk chromosomal abnormalities, the most potent ones are the deletions del(17p), t(4;14), and del(1p32)[190]. These deletions shorten progression free survival and overall survival. Other chromosomal changes do also impact survival, such as 1q gains,61 del(12p),30 or t(14;16)[191]. Secondary translocations and mutations acquired during disease progression include translocations targeting the MYC gene[192], gain of function mutations in oncogenes such as NRAS, KRAS, BRAF and CCND1, loss of function of tumor suppressors, e.g., P53, RB1, DIS3, CDKN2A and CDKN2C[193], and mutations in members of the NF-κB pathway[194] and STAT3[195]. The constitutional genotype of the tumor also plays a role in determining patient outcome[196]. Mutations in CDKN2A, a stabilizer of p53 increase sensitivity to chemotherapy[197]. Basmaci et al.[198] analyzed TNFα, NOS3 and MDR1 genetic status in MM patients and controls. There was no difference in the genetic status of MDR1. However, some genotypes were more common in the MM group compared to healthy controls suggesting a role in MM[198]. A SNP-array based study revealed that genetic abnormalities are of major prognostic value, representing 75% of the overall survival prediction[199]. One example is the BRAF V600E mutation, which provides information on the sensitivity to vemurafenib[200]. PSMB5 mutations caused BTZ-resistance in in vitro experiments in different leukemic cells[201]. Interestingly, no mutations were detected in the 5- to 6-fold BTZ-resistant 8226/BR7 MM cells[202], which supports the assumption that not only mutations of PSMB5 confer to resistance. Additionally there is a discrepancy between resistance mutations observed in lab conditions compared to clinical findings[203].
Role of epigenetics for drug resistance
Mutations of histone-modifying enzymes and other epigenetic regulators have been reported in numerous cancers including MM. Changes in the chromatin structure enable the rapid and reversible regulation of gene expression and subsequent biological events, including drug responses, without a heritable genetic alteration. This process, called epigenetic regulation, is accomplished through post-translational modifications of histone tails and DNA methylation. Alterations in these modifications entail various pathological conditions. In general, HATs tend to be inactivated in cancer cells. In MM, overexpression of the miRNA-106b-25 cluster down-regulates the expression of PCAF (p300/CBP-associated factor), a histone acetyltransferase involved in transcriptional control of TP53[204]. Recent investigations revealed that HDAC1, 2, 3 and 6 are highly expressed in MM cells and contribute to their malignant behavior[205]. Abnormal patterns of histone modifications are commonly observed in hematological malignancies. The MMSET-domain (multiple myeloma SET domain), potential oncogenic activity, is overexpressed in an aggressive form of MM carrying t(4;14), which is detected in 10%-15 % of all MM patients[129].
A particular target of significant clinical interest is the bromodomain and extra-terminal (BET) family of proteins, which includes BRD2, BRD3, BRD4, and the testis-specific BRDT. Bromodomains (BRDs) are protein domains that possess a high affinity for binding to acetylation motifs, including acetylated histone proteins within chromatin. The BET family of proteins bind to acetylated chromatin and regulate gene transcription. While BET proteins bind to enhancer sites throughout the genome, they are preferentially found at enhancer sites of oncogenes[206]. DNA methylation studies in cancer have concluded that malignant cells, including MM, are in general characterized by global DNA hypomethylation and hypermethylation of tumor suppressor genes[207]. In MM, global DNA hypomethylation correlates with disease progression and poor prognosis[208]. In addition, promoter DNA demethylation has been linked to drug resistance in MM by regulating the expression of the ATP-binding cassette (ABC) solute transporters. In response to chemotherapy, the expression of the ABCG2 gene is induced via demethylation, leading to increased drug efflux in MM cell lines and primary cells[142]. On the other hand, DNA hypermethylation of known tumor suppressor genes is commonly involved in the regulation of cell cycle, DNA repair, apoptosis, and key signaling pathways in MM.
miR-152, miR-10b-5p and miR-34c-3p have been defined as tumor suppressors, whose expression is impaired by DNA hypermethylation in MM. Induced expression of these microRNAs by DNA demethylating agents induces apoptosis, inhibits MM cell proliferation, and downregulates the expression of their putative oncogenic targets[209]. Furthermore, miR-155, miR-198, miR-135a, miR-200c, miR-663 and miR-483-5p are DNA hypermethylated in primary MM cells. Inhibiting DNA methylation using 5-azacytidine (AZA) reactivates microRNA expression. Importantly, ectopic expression of these microRNAs demonstrates anti-MM activities, thereby supporting their function as tumor suppressor microRNAs in MM[210]. Mutations in NSD1 and CREBBP genes are exclusively found in relapsed MM patients, suggesting that NSD1 may induces drug resistance[211,212]. Further studies evaluating the whole-genome DNA methylation, in parallel to locus specific DNA methylation patterns and functional studies on the consequences of global and regional demethylation are required for a better understanding of the role of DNA methylation in MM pathogenesis.
Treatment of refractory MM
Relapsed/refractory MM (RRMM) is defined as disease, which becomes non-responsive or progressive on therapy or within 60 days of the last treatment in patients, who had achieved a minimal response (MR) or better on prior therapy. While treatment is often indicated at relapse/progression of MM, a number of factors need to be considered before starting a therapy[213]. Relapsed MM has been defined based on objective laboratory and radiological criteria: ≥ 25% increase of the serum or urine monoclonal protein (M-protein) or ≥ 25% difference between involved and uninvolved serum free light chains from its nadir, respectively, or the development of new MM masses or hypercalcemia. In patients with non-secretory disease, relapse is defined as increase of the bone marrow plasma cells. In general, relapse of treatment has been defined as either the appearance or reappearance of one or more criteria or a rapid and consistent biochemical relapse[214]. The choice of therapy at relapse depends on a number of factors, including efficacy of and tolerance to previous therapy, number of previous treatments, time since relapse and aggressiveness, patient age and comorbidities[215]. Drug therapies at relapse frequently incorporate an immunomodulatory imide drug (IMiD), a class of immunomodulatory drugs that adjust immune responses containing an imide group like thalidomide, or a proteasome inhibitor in combination with dexamethasone [Table 1]. Results from phase III trials show that IMiDs are associated with significant improvements in overall response rate (ORR) and overall survival (OS) if used in combination with dexamethasone compared with dexamethasone alone[216,217]. Additionally, several studies have demonstrated increased efficacy, if different drug classes with distinct mechanisms of action are combined for the treatment of RRMM, and recent guidelines recommend combination therapies in this setting[218].
Overview of treatment options in RRMM
| Progress/refractory disease/recurrence | Treatment strategy |
|---|---|
| Patients who had one therapy + AST | BTZ/BTZ + liposomal Do with or without D |
| Patients who had one therapy, but were unsuitable for AST | BTZ/BTZ + liposomal Do with or without D |
| Patients who had one therapy | L/D |
| Patients with progressed MM or stadium II with progress or stadium III (according to Salmon Durie) | Bendamustine + P |
| Patients with two prior therapies (who already received BTZ and L) | Po + d |
Autologous stem cell transplantation
In autologous stem-cell transplantation (ASCT), stem cells are removed from a patient, stored, and reapplied later to that same person. The cells are stored in liquid nitrogen, until they are needed for the transplant. Then, the MM patient will be treated with high-dose chemotherapy or sometimes with radiation to efficiently kill the cancer cells. Afterwards, the stored stem cells are infused back into the patient’s blood. The survival of MM patients has greatly improved by using ASCT[219]. Sometimes two autologous transplantations are recommended, 6 to 12 months apart from each other. This tandem transplant approach is more efficient than single transplants, but it also causes more side effects[220].
Circulating plasma cells (CPCs) represent a risk factor for ASCT. CPCs have been detected in patients with MM at various stages of disease and have been associated with worse outcomes. The presence of CPCs at the time of autologous PBSC collection is a negative prognostic factor for risk of early relapse or death despite the advent of novel agents and maintenance strategies[221]. Circulating CD161-expressing cells, in addition to clinical risk factors, predict mucositis and infections in MM patients undergoing ASCT[222]. In 2017, the U.S. Food and Drug Administration (FDA) approved lenalidomide as maintenance therapy after ASCT for MM patients. The approval was based on evidence from two randomized, blinded trials of maintenance therapy with lenalidomide vs. placebo in MM patients, who underwent auto-HSCT along with a third trial on lenalidomide vs. no therapy. All trials demonstrated superior progression-free survival for the patients treated with lenalidomide[223]. Plerixafor, a hematopoietic stem cell mobilizer was combined with G-CSF and compared to placebo plus G-CSF in a clinical study to mobilize and collect CD34+ cells for autologous hematopoietic stem cell transplantation (HSCT). The addition of plerixafor to G-CSF for stem cell mobilization did not affect 5-year survival in patients with NHL or MM[224].
MM patients with a first relapse after prior ASCT benefit from a second ASCT as shown in a study comparing a second ASCT and lenalidomide maintenance therapy given at first relapse after prior ASCT with chemotherapy[225].
Novel agents
The treatment landscape for patients with RRMM is rapidly changing following the recent approval of drugs belonging to novel classes of agents such as histone deacetylase inhibitors, and two monoclonal antibodies. Furthermore, the addition of the second-generation IMiDs lenalidomide and pomalidomide and the second-generation proteasome inhibitors provides additional within-class treatment options for patients with RRMM.
Reversal of multidrug resistance would be an urgently needed, albeit not novel treatment approach. In several clinical trials, the effect of non-toxic substrates of P-g has been investigated to overcome MDR[226,227]. Valspodar (PSC833), an experimental cancer treatment and chemosensitizing drug is a derivative of cyclosporin D and was designed to prevent drug resistance to chemotherapeutics. A phase III study did not show any improvement by combining valspador with VAD (vincristine, vinblastine, doxorubicin and dexamethasone)[228]. The in vitro inhibition of miRNAs that correlated with melphalan sensitivity showed promising results in reversing melphalan resistance[229]. The new compound JNJ-26854165 induced cell death in an in vitro model through inhibition of cholesterol transport and degradation of the ABC-transporter ABCA1[230]. Differentiation therapy represents another strategy to reduce the stemness of CSCs. Withaferine A and withanolide D, two natural compounds were tested in MM-CSCs. They inhibited growth at nanomolar level and induced G2 cell cycle arrest as well as apoptotic cell death. Additionally, a remarkable morphological change was observed in MM CSCs[231,232]. In vitro trials with the telomerase inhibitor GRN163L demonstrated that resistant MM CSCs are susceptible to telomerase inhibition[233]. Ibrutinib, an oral covalent inhibitor of Bruton tyrosine kinase was tested in a phase II trial with patients suffering from RRMM. The drug regimen was either ibrutinib alone or in combination with dexamethasone. Response was observed in 51% of the patients treated with this combination regimen[234]. A large number of novel substances have been recently developed to target critical components in several epigenetic processes, particularly DNA methylation, histone acetylation and methylation. Some of these substances currently entered clinical practice[235]. Two small-molecule BET-inhibitor compounds, JQ1 and I-BET762, demonstrated a repression of c-Myc expression[206]. In a phase I b trial, the compound OTX015 was safely administered to older patients[236]. CARM1 is an arginine methyltransferase with diverse histone and non-histone substrates implicated in the regulation of cellular processes including transcriptional co-activation and RNA processing. CARM1 overexpression has been reported in multiple cancer types and has been shown to modulate oncogenic pathways in in vitro studies. EZM2302 (GSK3359088) is an inhibitor of CARM1 with enzymatic activity in biochemical assays (IC50 = 6 nmol/L) and with broad selectivity against other histone methyltransferases. Treatment of MM cell lines with EZM2302 led to inhibition of PABP1 and SMB methylation and cell stasis with IC50 values in the nanomolar range. Oral dosing of EZM2302 demonstrated dose-dependent in vivo CARM1 inhibition and anti-tumor activity in an MM xenograft model[237].
Novel drug combinations
The main aim of novel drug combinations is to overcome drug resistance. Cyclosporine A, a substrate of P-gp can be used to saturate P-gp in order to prevent efflux of active drugs. A study, which compared the combination of cyclosporine A with vincristine-doxorubicin-dexamethasone (VAD) with VAD alone, found no effect of cyclosporine A in the overall response rate and overall survival[238]. MM CSC-targeting therapies in combination with non-MM CSC therapies may offer a promising strategy for management of MM. A combination of 5-azacytidine and EPZ-6438 was used to treat IMiD-resistant cell lines. The combination re-sensitized resistant cells to both lenalidomide and pomalidomide. Simultaneous inhibition of DNA methyltransferases and EZH2 may lead to extensive epigenetic reprogramming, which allows MM cells to gain sensitivity to IMiDs[239]. The polycomb group (PcG) proteins BMI-1 of the polycomb repressive complex 1 (PRC1) possess oncogenic functions and are overexpressed in MM. PTC-209 exhibits synergistic and additive anti-MM activity, if combined with other epigenetic inhibitors targeting EZH2 and BET bromodomains[240]. Promising data have also been obtained with the proteasome-antibody combination, Elo-BTZ-dexamethasone. Elo-BTZ/d was evaluated in relapsed patients and achieved an ORR of 48% in the phase I trial[241]. A study observing the combination of daratumumab with BTZ/dexamethasone, BTZ/melphalan/prednisone, BTZ/thalidomide/dexamethasone, or pomalidomide/dexamethasone resulted in promising response rates. Daratumumab did not have a negative impact on stem cell mobilization. Phase III studies of daratumumab are planned or ongoing[242]. The majority of MM cells overexpresses syndecan-1 (CD138). CD138 is a heparin sulphate proteoglycan that controls tumor cell survival, growth, adhesion and bone cell differentiation in MM. Since CD138 is a specific surface antigen for MM cells and plasma cells in the bone marrow, it has been used for the purification of MM cells from clinical samples and in the classification of MM cells in gene expression profiling analyses. In addition, the use of specific antibodies targeting CD138 is also considered as novel treatment strategy. Indatuximab ravtansine (BT062) is monoclonal antibody-linked cytotoxic agent that specifically targets CD138-expressing cells. In vivo mouse models confirmed the activity of indatuximab ravtansine in combination with lenalidomide and lenalidomide/dexamethasone[243]. In contrast, a decrease in CD138 expression has been observed during the course of clinical treatment in some patients. While high expression of CD138 is not consistently observed in MM cells, the significance of decreased CD138 expression in MM cells remains unclear and could be a problem for CD138-targeted treatment[244]. A combination of dexamethasone with SST0001, a chemically modified heparin blocked tumor growth in vivo by disruption of the heparanase/syndecan-1 axis[245]. Another in vivo study observed the effect of a combination therapy of anti-CD138-IFNα14 fusion protein and BTZ. It cured tumors in animals[246].
Immunotherapy
An immunologic approach to MM offers promise for the treatment of this genetically heterogeneous disease, and patients with acquired drug resistance may particularly benefit from these therapies. MM is typically associated with severe immune dysfunction increasing both the risk of infections as well as other secondary malignancies. Malfunction of components of the innate and adoptive immune system in MM patients has been described[247]. In addition, MM cells specifically shield themselves from T cell responses by overexpressing protective molecules such as PD-L1, especially in the context of inflammation. Moreover, the pro-inflammatory cytokines IL-6 and IL-17 trigger MM cell growth and increase the number of immunosuppressive regulatory T cells (Tregs), resulting in a circulus vitiosus of immune paralysis and tumor progression[248]. Elotuzumab is a humanized IgG1 class monoclonal antibody targeting the cell surface protein signaling lymphocytic activation molecule F7 (syn. SLAMF7), a member of the immunoglobulin gene superfamily involved in immune cell activation. If evaluated in vitro, elotuzumab exerted its anti- MM activity primarily via antibody dependent cellular cytotoxicity (ADCC)[249]. Daratumumab (Darzalex, FDA approved since November 2015) reached a remarkable ORR of 36% including durable CRs beyond 1 year in a dose-escalating phase I trial. In a phase II trial focusing on dual (IMiDs and PI) refractory disease, single agent therapy with daratumumab still induced an objective response in 29% of patients including complete remissions[250]. A BAFF-receptor (human B-cell-activating factor receptor)-antibody induced potent ADCC against multiple subtypes of human lymphoma and leukemia[251]. Yang et al.[252] studied the use of an anti-ABCG2 monoclonal antibody in combination with paclitaxel nanoparticles in highly resistant MM cells. The combination led to a significant reduction in the tumor volume[252]. The human monoclonal antibody natalizumab was tested for the treatment of RRMM in a phase I/II trial[253]. Podar et al.[254] investigated its effect on MM cell proliferation. It inhibited VEGF secretion and angiogenesis and enhanced the anti-MM activity of BTZ and dexamethasone[254]. A number of IgG monoclonal antibodies specific to other MM surface targets are currently under investigation, either naked or with coupled immunotoxins. Some antibodies indeed showed objective responses in early clinical trials. However, if considering the ones with single agent activity (anti-CD40 lucatumumab, anti-CD138-immunotoxin indatuximab mavtansine, and anti-CD56-immunotoxin lorvotuzumab maytansine,) the response rates were limited to single cases, and dose-limiting toxicities made their further development as monotherapy unlikely[255].
The CS1 glycoprotein antigen could be potentially targeted with chimeric-antigen receptor (CAR) transduced T cells- with the goal to develop a clinical T cell therapy. CS1 is a glycoprotein expressed on the cell surface of nearly all MM cells. CARTS effectively redirected T cells to secrete cytokines, degranulate, and exhibit cytotoxic activity in response to MM cell lines and primary MM cells in vitro[256]. Another therapeutic strategy focuses on the activation of already existing tumor immunity, particularly on T cells. Blockage of PDL- 1 as well as inhibition of PD1 on T cells leads to the activation of tumor-infiltrating T cells specific for a variety of individual tumor antigens. Initial studies were disappointing, as no objective responses were seen[257]. Ongoing studies attempt to increase the activity of the expanded natural killer (NK) cell product and increase NK cell survival in vivo. In preclinical models, CAR engineered NK cells specific to SLAMF7 have also shown anti-MM activity[258].
Personalized treatment
MM is a biologically complex disease, with great heterogeneity in genetic aberrations as well as overall response and survival of patients. There is an extreme diversity of clinical manifestations ranging from more aggressive disease, characterized by disease resistance, to patients with relatively indolent disease, associated with longer survival. The aim of any risk stratification system is the optimization of therapeutic resources to improve depth and duration of response[259]. The Durie-Salmon Staging System (DSS) and the International Staging System (ISS) are the two most commonly used risk stratification models[260,261]. Both systems are based on tumor burden. Although this parameter has significant clinical utility, it is not devoid of limitations. DSS is almost redundant today considering low specificity of its factors (e.g., hemoglobin and serum creatinine) and the use of older techniques such as radiological skeletal survey and less predictability. Although convenient to use, ISS requires additional information from cytogenetics/fluorescence in situ hybridization (FISH) to make diagnosis more robust. By conventional techniques, cytogenetic abnormalities are detected in only 30%-40% of patients. This gross underestimation is the result of low mitotic index of malignant plasma cells, the telomeric locations of some of the chromosomal changes and the variable degree of bone marrow infiltration. With its ability to detect changes in interphase cells, FISH can overcome the problem of slowly or not proliferating cells and can be informative in almost all MM patients. The International Myeloma Working Group (IWMG) consensus has recommended a combination of ISS and FISH for risk stratification of MM patients. The European Myeloma Network strongly recommends testing for chromosome 1 abnormalities, as they have been shown to be correlated with a poor prognosis. More recent techniques such as high density comparative genomic hybridization, single-nucleotide polymorphism (SNP) arrays and gene expression profiling (GEP) are able to detect genomic aberrations in all MM patients. Comprehensive studies using microarray technology demonstrated associations of microRNAs with diagnosis, pathogenesis and prognosis of MM, indicating a potential role as novel biomarker[262].
With a focus on personalized therapy, the mSMART guidelines (stratification for multiple and risk-adapted therapy) have stratified MM patients into three groups to optimize therapy with targeted novel drugs: high risk, intermediate risk and low risk patients. Treatment of high risk patients focuses on combining various aggressive modalities including multi-agent induction chemotherapy (3- or 4-drug combinations), HDT/ASCT or even auto-allo tandem SCT, further consolidation and aggressive maintenance using 2-drug combinations including BTZ. Additionally, the use of newer proteasome inhibitors, second generation IMiDs and monoclonal antibodies should be considered as useful. In contrast, low risk patients with a better life expectancy are suitable for less toxic sequential therapy approach. On the other side, it is claimed that high dose multi drug-combination regimens (bortezomib, thalidomide, dexamethasone) will maximize patient survival without inducing any drug resistance[263]. The individualization of therapy does not end with initial induction and consolidation, but has to be incorporated into therapy of relapsed patients as well. Again, these novel agents alone may not confer the required treatment goals due to heterogeneity in tumor clones. Recurrent mutations can occur even late in the evolution of a tumor, thus necessitating combinations of molecularly targeted therapies with current approved treatment strategies.
The future of MM therapy should thus aim at integrated approaches to select optimized treatment strategies taking into account patient's clinical status, biochemical factors and targeting the genetic and/or epigenetic abnormalities present in individual MM tumors.
Conclusion
Overcoming drug resistance is one of the most important challenges in therapy of MM. Although most of the patients respond to the first treatment, their cancers become resistant with time. Genome-wide sequencing revealed a tremendous biological complexity of MM as characterized by a large intra- and inter-patient clonal heterogeneity and reflected a patient-to-patient variation concerning overall survival and response to treatment[264]. Cells exhibiting the MDR phenotype can become responsive to chemotherapy by treatment with MDR-reversing agents. These agents come from a diverse group of drugs that includes several membrane active agents, calmodulin antagonists, calcium channel blockers, local anesthetics and cyclosporine[265].
With the gain of knowledge on genetic and molecular changes in MM pathophysiology, many molecular targeted therapies including cell signaling targeted therapies are being developed for the treatment of relapsed o refractory MM. Additionally, the role of epigenetic aberrations such as DNA methylation, histone modifications and non-coding RNA expression in MM pathogenesis have steered MM management in new directions with the discovery of promising novel targets. Amongst these novel targeted therapies are inhibitors of histone deacetylase, Aurora kinase, PI3K/AKT/mTOR pathway, and cyclin-dependent kinase. MM microenvironment, cytokines, chemokines and growth factors forming the bone marrow milieu play a crucial role in growth, survival, adhesion, migration and apoptotic resistance of MM cells. Therapies targeting tumor microenvironment such as hypoxia, angiogenesis, integrins, CD44, C-X-C chemokine receptor type 4, and selectins are thus being explored in clinical studies. With the growing number of targets and newly developed drugs, it becomes a challenge to select the best treatment option for a patient. Therefore, guidelines for individualized treatment strategies will play an increasing role in therapy. The exploration of novel combination therapies based on genomic information with synergistic interactions to kill MM cells should be more in the focus of preclinical and clinical research to better cope with side effects and drug resistance phenomena and to exploit the full potential of individualized precision medicine.
Declarations
Authors’ contributionsConcept, design and intellectual content, manuscript editing and review: Efferth T
Literature search, literature analysis, manuscript preparation and editing: Nass J
Financial support and sponsorshipNass J obtained a PhD stipend of the Deutsche Forschungsgemeinschaft (DFG GRK 2015/1).
Conflicts of interestThe authors declare that there are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Leng Y, Hou J, Jin J, Zhang M, Ke X, Jiang B, Pan L, Yang L, Zhou F, Wang J, Wang Z, Liu L, Li W, Shen Z, Qiu L, Chang N, Li J, Liu J, Pang H, Meng H, Wei P, Jiang H, Liu Y, Zheng X, Yang S, Chen W. Circularly permuted TRAIL plus thalidomide and dexamethasone versus thalidomide and dexamethasone for relapsed/refractory multiple myeloma: a phase 2 study. Cancer Chemother Pharmacol 2017;79:1141-9.
2. Krishnan SR, Jaiswal R, Brown RD, Luk F, Bebawy M. Multiple myeloma and persistence of drug resistance in the age of novel drugs (Review). Int J Oncol 2016;49:33-50.
3. Iida S. Mechanisms of action and resistance for multiple myeloma novel drug treatments. Int J Hematol 2016;104:271-2.
4. Brenner H, Gondos A, Pulte D. Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood 2008;111:2521-6.
6. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci 2000;11:265-83.
7. Almond JB, Cohen GM. The proteasome: a novel target for cancer chemotherapy. Leukemia 2002;16:433-43.
8. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 2009;10:550-63.
9. Reits E, Griekspoor A, Neijssen J, Groothuis T, Jalink K, van Veelen P, Janssen H, Calafat J, Drijfhout JW, Neefjes J. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 2003;18:97-108.
10. Rock KL, York IA, Saric T, Goldberg AL. Protein degradation and the generation of MHC class I-presented peptides. Adv Immunol 2002;80:1-70.
11. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006;107:4907-16.
12. Kuhn DJ, Orlowski RZ. The immunoproteasome as a target in hematologic malignancies. Semin Hematol 2012;49:258-62.
13. Ettari R, Zappala M, Grasso S, Musolino C, Innao V, Allegra A. Immunoproteasome-selective and non-selective inhibitors: a promising approach for the treatment of multiple myeloma. Pharmacol Ther 2018;182:176-92.
14. Accardi F, Toscani D, Bolzoni M, Dalla Palma B, Aversa F, Giuliani N. Mechanism of action of bortezomib and the new proteasome inhibitors on myeloma cells and the bone microenvironment: impact on myeloma-induced alterations of bone remodeling. Biomed Res Int 2015;2015:172458.
15. Avet-Loiseau H, Bahlis NJ, Chng WJ, Masszi T, Viterbo L, Pour L, Ganly P, Palumbo A, Cavo M, Langer C, Pluta A, Nagler A, Kumar S, Ben-Yehuda D, Rajkumar SV, San-Miguel J, Berg D, Lin J, van de Velde H, Esseltine DL, di Bacco A, Moreau P, Richardson PG. Ixazomib significantly prolongs progression-free survival in high-risk relapsed/refractory myeloma patients. Blood 2017;130:2610-8.
16. Eek D, Krohe M, Mazar I, Horsfield A, Pompilus F, Friebe R, Shields AL. Patient-reported preferences for oral versus intravenous administration for the treatment of cancer: a review of the literature. Patient Prefer Adherence 2016;10:1609-21.
17. Infante JR, Mendelson DS, Burris HA 3rd, Bendell JC, Tolcher AW, Gordon MS, Gillenwater HH, Arastu-Kapur S, Wong HL, Papadopoulos KP. A first-in-human dose-escalation study of the oral proteasome inhibitor oprozomib in patients with advanced solid tumors. Invest New Drugs 2016;34:216-24.
18. Spencer A, Harrison S, Zonder J, Badros A, Laubach J, Bergin K, Khot A, Zimmerman T, Chauhan D, Levin N, MacLaren A, Reich SD, Trikha M, Richardson P. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): final study results. Br J Haematol 2018;180:41-51.
19. Levin N, Spencer A, Harrison SJ, Chauhan D, Burrows FJ, Anderson KC, Reich SD, Richardson PG, Trikha M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br J Haematol 2016;174:711-20.
20. Harrison SJ, Mainwaring P, Price T, Millward MJ, Padrik P, Underhill CR, Cannell PK, Reich SD, Trikha M, Spencer A. Phase I clinical trial of marizomib (NPI-0052) in patients with advanced malignancies including multiple myeloma: study NPI-0052-102 final results. Clin Cancer Res 2016;22:4559-66.
21. Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev 2017;36:561-84.
22. Shi Y, Gera J, Hu L, Hsu JH, Bookstein R, Li W, Lichtenstein A. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res 2002;62:5027-34.
23. Hideshima T, Chauhan D, Richardson P, Anderson KC. Identification and validation of novel therapeutic targets for multiple myeloma. J Clin Oncol 2005;23:6345-50.
24. Azab F, Vali S, Abraham J, Potter N, Muz B, de la Puente P, Fiala M, Paasch J, Sultana Z, Tyagi A, Abbasi T, Vij R, Azab AK. PI3KCA plays a major role in multiple myeloma and its inhibition with BYL719 decreases proliferation, synergizes with other therapies and overcomes stroma-induced resistance. Br J Haematol 2014;165:89-101.
25. Gera J, Lichtenstein A. The mammalian target of rapamycin pathway as a therapeutic target in multiple myeloma. Leuk Lymphoma 2011;52:1857-66.
26. Balsat M, Cornillon J. m-TOR inhibitors: biology and use in the treatment of haematological diseases. Bull Cancer 2011;98:935-43.
27. Hofmeister CC, Yang X, Pichiorri F, Chen P, Rozewski DM, Johnson AJ, Lee S, Liu Z, Garr CL, Hade EM, Ji J, Schaaf LJ, Benson DM Jr, Kraut EH, Hicks WJ, Chan KK, Chen CS, Farag SS, Grever MR, Byrd JC, Phelps MA. Phase I trial of lenalidomide and CCI-779 in patients with relapsed multiple myeloma: evidence for lenalidomide-CCI-779 interaction via P-glycoprotein. J Clin Oncol 2011;29:3427-34.
28. Yee AJ, Hari P, Marcheselli R, Mahindra AK, Cirstea DD, Scullen TA, Burke JN, Rodig SJ, Hideshima T, Laubach JP, Ghobrial IM, Schlossman RL, Munshi NC, Anderson KC, Weller EA, Richardson PG, Raje NS. Outcomes in patients with relapsed or refractory multiple myeloma in a phase I study of everolimus in combination with lenalidomide. Br J Haematol 2014;166:401-9.
29. Spencer A, Yoon SS, Harrison SJ, Morris SR, Smith DA, Brigandi RA, Gauvin J, Kumar R, Opalinska JB, Chen C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 2014;124:2190-5.
30. Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, Orford K, Aktan G, Ferron-Brady G, Ibrahim N, Gauvin J, Motwani M, Cornfeld M. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol 2015;75:183-9.
31. Trametinib and Akt Inhibitor GSK2141795 in Treating Patients With Relapsed or Refractory Multiple Myeloma. In: U.S. National Library of Medicine; 2018. ClinicalTrials.gov Identifier: NCT01989598.
32. Clinical Trials Using Akt inhibitor AZD5363. Available from: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/akt-inhibitor-azd5363 [Last accessed on 14 May 2018].
33. Oral ONC201 in Relapsed/Refractory Multiple Myeloma. In: U.S. National Library of Medicine; 2018. ClinicalTrials.gov Identifier: NCT02863991.
34. Richardson PG, Wolf J, Jakubowiak A, Zonder J, Lonial S, Irwin D, Densmore J, Krishnan A, Raje N, Bar M, Martin T, Schlossman R, Ghobrial IM, Munshi N, Laubach J, Allerton J, Hideshima T, Colson K, Poradosu E, Gardner L, Sportelli P, Anderson KC. Perifosine plus bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma previously treated with bortezomib: results of a multicenter phase I/II trial. J Clin Oncol 2011;29:4243-9.
35. Earwaker P, Anderson C, Willenbrock F, Harris AL, Protheroe AS, Macaulay VM. RAPTOR up-regulation contributes to resistance of renal cancer cells to PI3K-mTOR inhibition. PLoS One 2018;13:e0191890.
36. Xu J, Pfarr N, Endris V, Mai EK, Md Hanafiah NH, Lehners N, Penzel R, Weichert W, Ho AD, Schirmacher P, Goldschmidt H, Andrulis M, Raab MS. Molecular signaling in multiple myeloma: association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 2017;6:e337.
37. Chng WJ, Gonzalez-Paz N, Price-Troska T, Jacobus S, Rajkumar SV, Oken MM, Kyle RA, Henderson KJ, Van Wier S, Greipp P, Van Ness B, Fonseca R. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia 2008;22:2280-4.
38. Bisping G, Wenning D, Kropff M, Gustavus D, Muller-Tidow C, Stelljes M, Munzert G, Hilberg F, Roth GJ, Stefanic M, Volpert S, Mesters RM, Berdel WE, Kienast J. Bortezomib, dexamethasone, and fibroblast growth factor receptor 3-specific tyrosine kinase inhibitor in t(4;14) myeloma. Clin Cancer Res 2009;15:520-31.
39. Ramakrishnan V, Timm M, Haug JL, Kimlinger TK, Wellik LE, Witzig TE, Rajkumar SV, Adjei AA, Kumar S. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene 2010;29:1190-202.
40. Ramakrishnan V, Timm M, Haug JL, Kimlinger TK, Halling T, Wellik LE, Witzig TE, Rajkumar SV, Adjei AA, Kumar S. Sorafenib, a multikinase inhibitor, is effective in vitro against non-Hodgkin lymphoma and synergizes with the mTOR inhibitor rapamycin. Am J Hematol 2012;87:277-83.
41. Srkalovic G, Hussein MA, Hoering A, Zonder JA, Popplewell LL, Trivedi H, Mazzoni S, Sexton R, Orlowski RZ, Barlogie B. A phase II trial of BAY 43-9006 (sorafenib) (NSC-724772) in patients with relapsing and resistant multiple myeloma: SWOG S0434. Cancer Med 2014;3:1275-83.
42. Kumar SK, Jett J, Marks R, Richardson R, Quevedo F, Moynihan T, Croghan G, Markovic SN, Bible KC, Qin R, Tan A, Molina J, Kaufmann SH, Erlichman C, Adjei AA. Phase 1 study of sorafenib in combination with bortezomib in patients with advanced malignancies. Invest New Drugs 2013;31:1201-6.
43. Tai YT, Fulciniti M, Hideshima T, Song W, Leiba M, Li XF, Rumizen M, Burger P, Morrison A, Podar K, Chauhan D, Tassone P, Richardson P, Munshi NC, Ghobrial IM, Anderson KC. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood 2007;110:1656-63.
44. Ocio EM, Fernandez-Lazaro D, San-Segundo L, Lopez-Corral L, Corchete LA, Gutierrez NC, Garayoa M, Paino T, Garcia-Gomez A, Delgado M, Montero JC, Diaz-Rodriguez E, Mateos MV, Pandiella A, Couto S, Wang M, Bjorklund CC, San-Miguel JF. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia 2015;29:705-14.
45. Holkova B, Zingone A, Kmieciak M, Bose P, Badros AZ, Voorhees PM, Baz R, Korde N, Lin HY, Chen JQ, Herrmann M, Xi L, Raffeld M, Zhao X, Wan W, Tombes MB, Shrader E, Weir-Wiggins C, Sankala H, Hogan KT, Doyle A, Annunziata CM, Wellons M, Roberts JD, Sullivan D, Landgren O, Grant S. A phase II trial of AZD6244 (Selumetinib, ARRY-142886), an oral MEK1/2 inhibitor, in relapsed/refractory multiple myeloma. Clin Cancer Res 2016;22:1067-75.
46. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci 2004;117:1281-3.
47. Beldi-Ferchiou A, Skouri N, Ben Ali C, Safra I, Abdelkefi A, Ladeb S, Mrad K, Ben Othman T, Ben Ahmed M. Abnormal repression of SHP-1, SHP-2 and SOCS-1 transcription sustains the activation of the JAK/STAT3 pathway and the progression of the disease in multiple myeloma. PLoS One 2017;12:e0174835.
48. Ramakrishnan V, Kimlinger T, Haug J, Timm M, Wellik L, Halling T, Pardanani A, Tefferi A, Rajkumar SV, Kumar S. TG101209, a novel JAK2 inhibitor, has significant in vitro activity in multiple myeloma and displays preferential cytotoxicity for CD45+ myeloma cells. Am J Hematol 2010;85:675-86.
49. de Oliveira MB, Fook-Alves VL, Eugenio AIP, Fernando RC, Sanson LFG, de Carvalho MF, Braga WMT, Davies FE, Colleoni GWB. Anti-myeloma effects of ruxolitinib combined with bortezomib and lenalidomide: a rationale for JAK/STAT pathway inhibition in myeloma patients. Cancer Lett 2017;403:206-15.
50. Sagawa M, Tabayashi T, Kimura Y, Tomikawa T, Nemoto-Anan T, Watanabe R, Tokuhira M, Ri M, Hashimoto Y, Iida S, Kizaki M. TM-233, a novel analog of 1'-acetoxychavicol acetate, induces cell death in myeloma cells by inhibiting both JAK/STAT and proteasome activities. Cancer Sci 2015;106:438-46.
51. Sagawa M, Nakazato T, Uchida H, Ikeda Y, Kizaki M. Cantharidin induces apoptosis of human multiple myeloma cells via inhibition of the JAK/STAT pathway. Cancer Sci 2008;99:1820-6.
52. Nakaya A, Sagawa M, Muto A, Uchida H, Ikeda Y, Kizaki M. The gold compound auranofin induces apoptosis of human multiple myeloma cells through both down-regulation of STAT3 and inhibition of NF-kappaB activity. Leuk Res 2011;35:243-9.
53. Le Gouill S, Pellat-Deceunynck C, Harousseau JL, Rapp MJ, Robillard N, Bataille R, Amiot M. Farnesyl transferase inhibitor R115777 induces apoptosis of human myeloma cells. Leukemia 2002;16:1664-7.
54. Li J, Favata M, Kelley JA, Caulder E, Thomas B, Wen X, Sparks RB, Arvanitis A, Rogers JD, Combs AP, Vaddi K, Solomon KA, Scherle PA, Newton R, Fridman JS. INCB16562, a JAK1/2 selective inhibitor, is efficacious against multiple myeloma cells and reverses the protective effects of cytokine and stromal cell support. Neoplasia 2010;12:28-38.
55. Jin Z, Zhou S, Zhang Y, Ye H, Jiang S, Yu K, Ma Y. Lycorine induces cell death in MM by suppressing Janus kinase/signal transducer and activator of transcription via inducing the expression of SOCS1. Biomed Pharmacother 2016;84:1645-53.
56. Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, Bornmann W, Bromberg J. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res 2006;66:9714-21.
57. Amit-Vazina M, Shishodia S, Harris D, Van Q, Wang M, Weber D, Alexanian R, Talpaz M, Aggarwal BB, Estrov Z. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br J Cancer 2005;93:70-80.
58. Safety and Efficacy of Atiprimod for Patients With Refractory Multiple Myeloma. In: U.S. National Library of Medicine; 2018. ClinicalTrials.gov Identifier: NCT00086216. .
59. Kampan NC, Xiang SD, McNally OM, Stephens AN, Quinn MA, Plebanski M. Immunotherapeutic Interleukin-6 or Interleukin-6 receptor blockade in cancer: challenges and opportunities. Curr Med Chem 2017; doi: 10.2174/0929867324666170712160621. doi: 10.2174/0929867324666170712160621
60. Orlowski RZ, Gercheva L, Williams C, Sutherland H, Robak T, Masszi T, Goranova-Marinova V, Dimopoulos MA, Cavenagh JD, Spicka I, Maiolino A, Suvorov A, Blade J, Samoylova O, Puchalski TA, Reddy M, Bandekar R, van de Velde H, Xie H, Rossi JF. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am J Hematol 2015;90:42-9.
61. San-Miguel J, Blade J, Shpilberg O, Grosicki S, Maloisel F, Min CK, Polo Zarzuela M, Robak T, Prasad SV, Tee Goh Y, Laubach J, Spencer A, Mateos MV, Palumbo A, Puchalski T, Reddy M, Uhlar C, Qin X, van de Velde H, Xie H, Orlowski RZ. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood 2014;123:4136-42.
62. Follin-Arbelet V, Torgersen ML, Naderi EH, Misund K, Sundan A, Blomhoff HK. Death of multiple myeloma cells induced by cAMP-signaling involves downregulation of Mcl-1 via the JAK/STAT pathway. Cancer Lett 2013;335:323-31.
63. Ko JH, Ho Baek S, Nam D, Chung WS, Lee SG, Lee J, Mo Yang W, Um JY, Seok Ahn K. 3-Formylchromone inhibits proliferation and induces apoptosis of multiple myeloma cells by abrogating STAT3 signaling through the induction of PIAS3. Immunopharmacol Immunotoxicol 2016;38:334-43.
64. Monaghan KA, Khong T, Burns CJ, Spencer A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia 2011;25:1891-9.
65. Che Y, Hou S, Kang Z, Lin Q. Serenoa repens induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of STAT 3 signaling. Oncol Rep 2009;22:377-83.
66. Pandey MK, Sung B, Ahn KS, Aggarwal BB. Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol Pharmacol 2009;75:525-33.
67. Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, Chauhan D, Anderson KC, Frank DA. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood 2008;112:5095-102.
68. De Vos J, Jourdan M, Tarte K, Jasmin C, Klein B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. Br J Haematol 2000;109:823-8.
70. Grainger S, Traver D, Willert K. Wnt signaling in hematological malignancies. Prog Mol Biol Transl Sci 2018;153:321-41.
71. Cheng Q, Liu L, Fu Y, Zhang Y, Yang Y, Liu J. RhPDCD5 combined with dexamethasone increases antitumor activity in multiple myeloma partially via inhibiting the Wnt signalling pathway. Clin Exp Pharmacol Physiol 2018;45:140-5.
72. Wang JH, Zhang Y, Li HY, Liu YY, Sun T. Dickkopf-1 negatively regulates the expression of osteoprotegerin, a key osteoclastogenesis inhibitor, by sequestering Lrp6 in primary and metastatic lytic bone lesions. Medicine (Baltimore) 2016;95:e3767.
73. Zhou F, Meng S, Song H, Claret FX. Dickkopf-1 is a key regulator of myeloma bone disease: opportunities and challenges for therapeutic intervention. Blood Rev 2013;27:261-7.
74. Study of BHQ880 in Patients With High Risk Smoldering Multiple Myeloma. In: U.S. National Library of Medicine; 2018. ClinicalTrials.gov Identifier: NCT01302886.
75. Terpos E, Christoulas D, Gavriatopoulou M, Dimopoulos MA. Mechanisms of bone destruction in multiple myeloma. Eur J Cancer Care (Engl) 2017;26:12761.
76. McDonald MM, Reagan MR, Youlten SE, Mohanty ST, Seckinger A, Terry RL, Pettitt JA, Simic MK, Cheng TL, Morse A, Le LMT, Abi-Hanna D, Kramer I, Falank C, Fairfield H, Ghobrial IM, Baldock PA, Little DG, Kneissel M, Vanderkerken K, Bassett JHD, Williams GR, Oyajobi BO, Hose D, Phan TG, Croucher PI. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017;129:3452-64.
77. Bjorklund CC, Ma W, Wang ZQ, Davis RE, Kuhn DJ, Kornblau SM, Wang M, Shah JJ, Orlowski RZ. Evidence of a role for activation of Wnt/beta-catenin signaling in the resistance of plasma cells to lenalidomide. J Biol Chem 2011;286:11009-20.
78. Butrym A, Rybka J, Lacina P, Gebura K, Frontkiewicz D, Bogunia-Kubik K, Mazur G. Polymorphisms within beta-catenin encoding gene affect multiple myeloma development and treatment. Leuk Res 2015;39:1462-6.
79. Demchenko YN, Kuehl WM. A critical role for the NFkB pathway in multiple myeloma. Oncotarget 2010;1:59-68.
80. Chim CS. Updated survivals and prognostic factor analysis in myeloma treated by a staged approach use of bortezomib/thalidomide/dexamethasone in transplant eligible patients. J Transl Med 2010;8:124.
81. Chng WJ, Kumar S, Vanwier S, Ahmann G, Price-Troska T, Henderson K, Chung TH, Kim S, Mulligan G, Bryant B, Carpten J, Gertz M, Rajkumar SV, Lacy M, Dispenzieri A, Kyle R, Greipp P, Bergsagel PL, Fonseca R. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res 2007;67:2982-9.
82. Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007;12:131-44.
83. Bolzoni M, Toscani D, Storti P, Marchica V, Costa F, Giuliani N. Possible targets to treat myeloma-related osteoclastogenesis. Expert Rev Hematol 2018;11:325-36.
84. Terpos E, Efstathiou E, Christoulas D, Roussou M, Katodritou E, Dimopoulos MA. RANKL inhibition: clinical implications for the management of patients with multiple myeloma and solid tumors with bone metastases. Expert Opin Biol Ther 2009;9:465-79.
85. Raje N, Vadhan-Raj S, Willenbacher W, Terpos E, Hungria V, Spencer A, Alexeeva Y, Facon T, Stewart AK, Feng A, Braun A, Balakumaran A, Roodman GD. Evaluating results from the multiple myeloma patient subset treated with denosumab or zoledronic acid in a randomized phase 3 trial. Blood Cancer J 2016;6:e378.
86. Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, Harousseau JL, Lipton A, Mariette X, Williams CD, Nakanishi A, Holloway D, Martin SW, Dunstan CR, Bekker PJ. A phase I study of AMGN-0007, a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer 2003;97:887-92.
87. Terpos E, Mihou D, Szydlo R, Tsimirika K, Karkantaris C, Politou M, Voskaridou E, Rahemtulla A, Dimopoulos MA, Zervas K. The combination of intermediate doses of thalidomide with dexamethasone is an effective treatment for patients with refractory/relapsed multiple myeloma and normalizes abnormal bone remodeling, through the reduction of sRANKL/osteoprotegerin ratio. Leukemia 2005;19:1969-76.
88. Tsubaki M, Takeda T, Yoshizumi M, Ueda E, Itoh T, Imano M, Satou T, Nishida S. RANK-RANKL interactions are involved in cell adhesion-mediated drug resistance in multiple myeloma cell lines. Tumour Biol 2016;37:9099-110.
89. Santo L, Siu KT, Raje N. Targeting cyclin-dependent kinases and cell cycle progression in human cancers. Semin Oncol 2015;42:788-800.
90. Casimiro MC, Velasco-Velazquez M, Aguirre-Alvarado C, Pestell RG. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opin Investig Drugs 2014;23:295-304.
91. Ramakrishnan VG, Kumar SK. Inhibitors of the cyclin-dependent kinase and PIM kinase pathways in the treatment of myeloma. Cancer J 2016;22:7-11.
92. MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res 2005;65:5399-407.
93. Hofmeister CC, Poi M, Bowers MA, Zhao W, Phelps MA, Benson DM, Kraut EH, Farag S, Efebera YA, Sexton J, Lin TS, Grever M, Byrd JC. A phase I trial of flavopiridol in relapsed multiple myeloma. Cancer Chemother Pharmacol 2014;73:249-57.
94. Holkova B, Perkins EB, Ramakrishnan V, Tombes MB, Shrader E, Talreja N, Wellons MD, Hogan KT, Roodman GD, Coppola D, Kang L, Dawson J, Stuart RK, Peer C, Figg WD Sr, Kolla S, Doyle A, Wright J, Sullivan DM, Roberts JD, Grant S. Phase I trial of bortezomib (PS-341; NSC 681239) and alvocidib (flavopiridol; NSC 649890) in patients with recurrent or refractory B-cell neoplasms. Clin Cancer Res 2011;17:3388-97.
95. Fandy TE, Ross DD, Gore SD, Srivastava RK. Flavopiridol synergizes TRAIL cytotoxicity by downregulation of FLIPL. Cancer Chemother Pharmacol 2007;60:313-9.
96. Pei XY, Dai Y, Grant S. The small-molecule Bcl-2 inhibitor HA14-1 interacts synergistically with flavopiridol to induce mitochondrial injury and apoptosis in human myeloma cells through a free radical-dependent and Jun NH2-terminal kinase-dependent mechanism. Mol Cancer Ther 2004;3:1513-24.
97. Niesvizky R, Badros AZ, Costa LJ, Ely SA, Singhal SB, Stadtmauer EA, Haideri NA, Yacoub A, Hess G, Lentzsch S, Spicka I, Chanan-Khan AA, Raab MS, Tarantolo S, Vij R, Zonder JA, Huang X, Jayabalan D, Di Liberto M, Huang X, Jiang Y, Kim ST, Randolph S, Chen-Kiang S. Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leuk Lymphoma 2015;56:3320-8.
98. Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, Fruth B, Roy V, Erlichman C, Stewart AK, Mayo Phase 2 Consortium. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015;125:443-8.
99. Feng R, Li S, Lu C, Andreas C, Stolz DB, Mapara MY, Lentzsch S. Targeting the microtubular network as a new antimyeloma strategy. Mol Cancer Ther 2011;10:1886-96.
100. Maes A, Menu E, Veirman K, Maes K, Vand Erkerken K, De Bruyne E. The therapeutic potential of cell cycle targeting in multiple myeloma. Oncotarget 2017;8:90501-20.
101. Kyle RA, Jacobus S, Friedenberg WR, Slabber CF, Rajkumar SV, Greipp PR. The treatment of multiple myeloma using vincristine, carmustine, melphalan, cyclophosphamide, and prednisone (VBMCP) alternating with high-dose cyclophosphamide and alpha(2)beta interferon versus VBMCP: results of a phase III Eastern Cooperative Oncology Group Study E5A93. Cancer 2009;115:2155-64.
102. Dimopoulos MA, Arbuck S, Huber M, Weber D, Luckett R, Delasalle K, Alexanian R. Primary therapy of multiple myeloma with paclitaxel (taxol). Ann Oncol 1994;5:757-9.
103. Miller HJ, Leong T, Khandekar JD, Greipp PR, Gertz MA, Kyle RA. Paclitaxel as the initial treatment of multiple myeloma: an Eastern Cooperative Oncology Group Study (E1A93). Am J Clin Oncol 1998;21:553-6.
104. Kawano T, Agata N, Kharbanda S, Avigan D, Kufe D. A novel isocoumarin derivative induces mitotic phase arrest and apoptosis of human multiple myeloma cells. Cancer Chemother Pharmacol 2007;59:329-35.
105. Maginn EN, Browne PV, Hayden P, Vandenberghe E, MacDonagh B, Evans P, Goodyer M, Tewari P, Campiani G, Butini S, Williams DC, Zisterer DM, Lawler MP, McElligott AM. PBOX-15, a novel microtubule targeting agent, induces apoptosis, upregulates death receptors, and potentiates TRAIL-mediated apoptosis in multiple myeloma cells. Br J Cancer 2011;104:281-9.
106. Spagnuolo PA, Hu J, Hurren R, Wang X, Gronda M, Sukhai MA, Di Meo A, Boss J, Ashali I, Beheshti Zavareh R, Fine N, Simpson CD, Sharmeen S, Rottapel R, Schimmer AD. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood 2010;115:4824-33.
107. Pandey MK, Gowda K, Sung SS, Abraham T, Budak-Alpdogan T, Talamo G, Dovat S, Amin S. A novel dual inhibitor of microtubule and Bruton's tyrosine kinase inhibits survival of multiple myeloma and osteoclastogenesis. Exp Hematol 2017;53:31-42.
108. Rozic G, Paukov L, Jakubikova J, Ben-Shushan D, Duek A, Leiba A, Avigdor A, Nagler A, Leiba M. The novel compound STK405759 is a microtubule-targeting agent with potent and selective cytotoxicity against multiple myeloma in vitro and in vivo. Oncotarget 2016;7:62572-84.
109. Liu M, Aneja R, Liu C, Sun L, Gao J, Wang H, Dong JT, Sarli V, Giannis A, Joshi HC, Zhou J. Inhibition of the mitotic kinesin Eg5 up-regulates Hsp70 through the phosphatidylinositol 3-kinase/Akt pathway in multiple myeloma cells. J Biol Chem 2006;281:18090-7.
110. Hernandez-Garcia S, San-Segundo L, Gonzalez-Mendez L, Corchete LA, Misiewicz-Krzeminska I, Martin-Sanchez M, Lopez-Iglesias AA, Algarin EM, Mogollon P, Diaz-Tejedor A, Paino T, Tunquist B, Mateos MV, Gutierrez NC, Diaz-Rodriguez E, Garayoa M, Ocio EM. The kinesin spindle protein inhibitor filanesib enhances the activity of pomalidomide and dexamethasone in multiple myeloma. Haematologica 2017;102:2113-24.
111. Shah JJ, Kaufman JL, Zonder JA, Cohen AD, Bensinger WI, Hilder BW, Rush SA, Walker DH, Tunquist BJ, Litwiler KS, Ptaszynski M, Orlowski RZ, Lonial S. A phase 1 and 2 study of Filanesib alone and in combination with low-dose dexamethasone in relapsed/refractory multiple myeloma. Cancer 2017;123:4617-30.
112. Chari A, Htut M, Zonder JA, Fay JW, Jakubowiak AJ, Levy JB, Lau K, Burt SM, Tunquist BJ, Hilder BW, Rush SA, Walker DH, Ptaszynski M, Kaufman JL. A phase 1 dose-escalation study of filanesib plus bortezomib and dexamethasone in patients with recurrent/refractory multiple myeloma. Cancer 2016;122:3327-35.
113. Song IS, Jeong YJ, Nyamaa B, Jeong SH, Kim HK, Kim N, Ko KS, Rhee BD, Han J. KSP inhibitor SB743921 induces death of multiple myeloma cells via inhibition of the NF-kappaB signaling pathway. BMB Rep 2015;48:571-6.
114. Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J. Aurora kinases: novel therapy targets in cancers. Oncotarget 2017;8:23937-54.
115. Shi Y, Reiman T, Li W, Maxwell CA, Sen S, Pilarski L, Daniels TR, Penichet ML, Feldman R, Lichtenstein A. Targeting aurora kinases as therapy in multiple myeloma. Blood 2007;109:3915-21.
116. Aurora A Kinase Inhibitor MLN8237 and Bortezomib in Treating Patients With Relapsed or Refractory Multiple Myeloma. In: U.S. National Institutes of Health; 2018. ClinicalTrials.gov Identifier: NCT01034553.
117. Hay AE, Murugesan A, DiPasquale AM, Kouroukis T, Sandhu I, Kukreti V, Bahlis NJ, Lategan J, Reece DE, Lyons JF, Sederias J, Xu H, Powers J, Seymour LK, Reiman T. A phase II study of AT9283, an aurora kinase inhibitor, in patients with relapsed or refractory multiple myeloma: NCIC clinical trials group IND.191. Leuk Lymphoma 2016;57:1463-6.
118. Abdi J, Rastgoo N, Li L, Chen W, Chang H. Role of tumor suppressor p53 and micro-RNA interplay in multiple myeloma pathogenesis. J Hematol Oncol 2017;10:169.
119. Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017;8:8921-46.
120. Gillardin PS, Descamps G, Maiga S, Tessoulin B, Djamai H, Lucani B, Chiron D, Moreau P, Le Gouill S, Amiot M, Pellat-Deceunynck C, Moreau-Aubry A. Decitabine and melphalan fail to reactivate p73 in p53 deficient myeloma cells. Int J Mol Sci 2017;19:E40.
121. Hideshima T, Cottini F, Nozawa Y, Seo HS, Ohguchi H, Samur MK, Cirstea D, Mimura N, Iwasawa Y, Richardson PG, Munshi NC, Chauhan D, Massefski W, Utsugi T, Dhe-Paganon S, Anderson KC. p53-related protein kinase confers poor prognosis and represents a novel therapeutic target in multiple myeloma. Blood 2017;129:1308-19.
122. Liu N, Li S, Wu N, Cho KS. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017;8:89315-25.
123. Mihaila RG. From a better understanding of the mechanisms of action of histone deacetylases inhibitors to the progress of the treatment of malignant lymphomas and plasma cell myeloma. Recent Pat Anticancer Drug Discov 2017;12:283-95.
124. Raedler LA. Farydak (Panobinostat): first HDAC inhibitor approved for patients with relapsed multiple myeloma. Am Health Drug Benefits 2016;9:84-7.
125. Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, Harris C, Zwiebel J, Wright JJ, Espinoza-Delgado I, Baer MR, Holleran JL, Egorin MJ, Grant S. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res 2009;15:5250-7.
126. Deming DA, Ninan J, Bailey HH, Kolesar JM, Eickhoff J, Reid JM, Ames MM, McGovern RM, Alberti D, Marnocha R, Espinoza-Delgado I, Wright J, Wilding G, Schelman WR. A phase I study of intermittently dosed vorinostat in combination with bortezomib in patients with advanced solid tumors. Invest New Drugs 2014;32:323-9.
127. Cho H, Yoon DH, Kim KP, Bae KS, Kim WS, Eom HS, Kim JS, Hong JY, Kim SJ, Lee H, Kim SJ, Suh C. Phase I study of CKD-581, a pan-histone deacetylase inhibitor, in patients with lymphoma or multiple myeloma refractory to standard therapy. Invest New Drugs 2018; doi: 10.1007/s10637-018-0582-0. doi: 10.1007/s10637-018-0582-0.
128. Klappe K, Hinrichs JW, Kroesen BJ, Sietsma H, Kok JW. MRP1 and glucosylceramide are coordinately over expressed and enriched in rafts during multidrug resistance acquisition in colon cancer cells. Int J Cancer 2004;110:511-22.
129. Furukawa Y, Kikuchi J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int J Hematol 2016;104:281-92.
130. Raaijmakers MH. ATP-binding-cassette transporters in hematopoietic stem cells and their utility as therapeutical targets in acute and chronic myeloid leukemia. Leukemia 2007;21:2094-102.
131. Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009;323:1718-22.
132. Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc Natl Acad Sci U S A 2007;104:19005-10.
133. Ferreira RJ, Ferreira MJ, Dos Santos DJ. Insights on P-glycoprotein's efflux mechanism obtained by molecular dynamics simulations. J Chem Theory Comput 2012;8:1853-64.
134. Abraham J, Salama NN, Azab AK. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk Lymphoma 2015;56:26-33.
135. Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, Oerlemans R, Chan ET, Kirk CJ, Peters GJ, van der Heijden JW, de Gruijl TD, Scheper RJ, Jansen G. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 2012;341:174-82.
136. O'Connor R, Ooi MG, Meiller J, Jakubikova J, Klippel S, Delmore J, Richardson P, Anderson K, Clynes M, Mitsiades CS, O'Gorman P. The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother Pharmacol 2013;71:1357-68.
137. Dalton WS, Grogan TM, Meltzer PS, Scheper RJ, Durie BG, Taylor CW, Miller TP, Salmon SE. Drug-resistance in multiple myeloma and non-Hodgkin's lymphoma: detection of P-glycoprotein and potential circumvention by addition of verapamil to chemotherapy. J Clin Oncol 1989;7:415-24.
138. Grogan T, Dalton W, Rybski J, Spier C, Meltzer P, Richter L, Gleason M, Pindur J, Cline A, Scheper R. Optimization of immunocytochemical P-glycoprotein assessment in multidrug-resistant plasma cell myeloma using three antibodies. Lab Invest 1990;63:815-24.
139. Fonti R, Del Vecchio S, Zannetti A, De Renzo A, Catalano L, Pace L, Rotoli B, Salvatore M. Functional imaging of multidrug resistant phenotype by 99mTc-MIBI scan in patients with multiple myeloma. Cancer Biother Radiopharm 2004;19:165-70.
140. Fallahi B, Beiki D, Mousavi SA, Gholamrezanezhad A, Eftekhari M, Fard-Esfahani A, Alimoghaddam K, Mirpour S, Eskandarian A, Saghari M. 99mTc-MIBI whole body scintigraphy and P-glycoprotein for the prediction of multiple drug resistance in multiple myeloma patients. Hell J Nucl Med 2009;12:255-9.
141. Marie JP. P-glycoprotein in adult hematologic malignancies. Hematol Oncol Clin North Am 1995;9:239-49.
142. Turner JG, Gump JL, Zhang C, Cook JM, Marchion D, Hazlehurst L, Munster P, Schell MJ, Dalton WS, Sullivan DM. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006;108:3881-9.
143. Advani AS, Shadman M, Ali-Osman F, Barker A, Rybicki L, Kalaycio M, Sekeres MA, de Castro CM, Diehl LF, Moore JO, Beaven A, Copelan E, Sobecks R, Talea P, Rizzieri DA. A phase II trial of gemcitabine and mitoxantrone for patients with acute myeloid leukemia in first relapse. Clin Lymphoma Myeloma Leuk 2010;10:473-6.
144. Yin G, Xiao Z, Ni Y, Qu X, Wu H, Lu H, Qian S, Chen L, Li J, Qiu H, Miao K. Association of MDR1 single-nucleotide polymorphisms and haplotype variants with multiple myeloma in Chinese Jiangsu Han population. Tumour Biol 2016;37:9549-54.
145. Martino A, Campa D, Buda G, Sainz J, Garcia-Sanz R, Jamroziak K, Reis RM, Weinhold N, Jurado M, Rios R, Szemraj-Rogucka Z, Marques H, Szemraj J, Stein A, Kumar R, Orciuolo E, Gemignani F, Landi S, Goldschmidt H, Petrini M, Dumontet C, Canzian F, Rossi AM. Polymorphisms in xenobiotic transporters ABCB1, ABCG2, ABCC2, ABCC1, ABCC3 and multiple myeloma risk: a case-control study in the context of the International Multiple Myeloma rESEarch (IMMEnSE) consortium. Leukemia 2012;26:1419-22.
146. Drain S, Catherwood MA, Bjourson AJ, Drake MB, Kettle PJ, Alexander HD. Neither P-gp SNP variants, P-gp expression nor functional P-gp activity predicts MDR in a preliminary study of plasma cell myeloma. Cytometry B Clin Cytom 2012;82:229-37.
147. Schaich M, Soucek S, Thiede C, Ehninger G, Illmer T; SHG AML96 Study Group. MDR1 and MRP1 gene expression are independent predictors for treatment outcome in adult acute myeloid leukaemia. Br J Haematol 2005;128:324-32.
148. Ao L, Wu Y, Kim D, Jang ER, Kim K, Lee DM, Kim KB, Lee W. Development of peptide-based reversing agents for p-glycoprotein-mediated resistance to carfilzomib. Mol Pharm 2012;9:2197-205.
149. Lilienbaum A. Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol 2013;4:1-26.
150. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009;15:5308-16.
151. Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Res 2009;69:4415-23.
152. Ding WX, Ni HM, Gao W, Chen X, Kang JH, Stolz DB, Liu J, Yin XM. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Cancer Ther 2009;8:2036-45.
153. Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol 2017;10:67.
154. Podar K, Raab MS, Zhang J, McMillin D, Breitkreutz I, Tai YT, Lin BK, Munshi N, Hideshima T, Chauhan D, Anderson KC. Targeting PKC in multiple myeloma: in vitro and in vivo effects of the novel, orally available small-molecule inhibitor enzastaurin (LY317615.HCl). Blood 2007;109:1669-77.
155. Raab MS, Breitkreutz I, Tonon G, Zhang J, Hayden PJ, Nguyen T, Fruehauf JH, Lin BK, Chauhan D, Hideshima T, Munshi NC, Anderson KC, Podar K. Targeting PKC: a novel role for beta-catenin in ER stress and apoptotic signaling. Blood 2009;113:1513-21.
156. Yang Y, Chen Y, Saha MN, Chen J, Evans K, Qiu L, Reece D, Chen GA, Chang H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015;29:715-26.
157. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730-7.
158. Franqui-Machin R, Wendlandt EB, Janz S, Zhan F, Tricot G. Cancer stem cells are the cause of drug resistance in multiple myeloma: fact or fiction? Oncotarget 2015;6:40496-506.
159. Hu JS, Huang X, Huang YD, Lu YY, Lu QY. Effect of Hedgehog signaling pathway abnormality on chemothe-rapeutic resistance of multiple myeloma. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2017;25:465-70. (in Chinese)
160. Zhao JJ, Lin J, Zhu D, Wang X, Brooks D, Chen M, Chu ZB, Takada K, Ciccarelli B, Admin S, Tao J, Tai YT, Treon S, Pinkus G, Kuo WP, Hideshima T, Bouxsein M, Munshi N, Anderson K, Carrasco R. miR-30-5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/beta-catenin/BCL9 pathway. Cancer Res 2014;74:1801-13.
161. Tang B, Xu A, Xu J, Huang H, Chen L, Su Y, Zhang L, Li J, Fan F, Deng J, Tang L, Sun C, Hu Y. MicroRNA-324-5p regulates stemness, pathogenesis and sensitivity to bortezomib in multiple myeloma cells by targeting hedgehog signaling. Int J Cancer 2018;142:109-20.
162. Xu Z, Huang C, Hao D. MicroRNA-1271 inhibits proliferation and promotes apoptosis of multiple myeloma cells through inhibiting smoothened-mediated Hedgehog signaling pathway. Oncol Rep 2017;37:1261-9.
163. Yang Y, Shi J, Tolomelli G, Xu H, Xia J, Wang H, Zhou W, Zhou Y, Das S, Gu Z, Levasseur D, Zhan F, Tricot G. RARalpha2 expression confers myeloma stem cell features. Blood 2013;122:1437-47.
164. Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000;287:1804-8.
165. Januchowski R, Wojtowicz K, Zabel M. The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed Pharmacother 2013;67:669-80.
166. Saltarella I, Lamanuzzi A, Reale A, Vacca A, Ria R. Identify multiple myeloma stem cells: Utopia? World J Stem Cells 2015;7:84-95.
167. Mirandola L, Apicella L, Colombo M, Yu Y, Berta DG, Platonova N, Lazzari E, Lancellotti M, Bulfamante G, Cobos E, Chiriva-Internati M, Chiaramonte R. Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia 2013;27:1558-66.
168. Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, Lin BK, Gupta D, Shima Y, Chauhan D, Mitsiades C, Raje N, Richardson P, Anderson KC. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001;98:428-35.
169. Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res 2008;14:2519-26.
170. Bianchi G, Kumar S, Ghobrial IM, Roccaro AM. Cell trafficking in multiple myeloma. Open J Hematol 2012;3:4.
171. Bar-Natan M, Stroopinsky D, Luptakova K, Coll MD, Apel A, Rajabi H, Pyzer AR, Palmer K, Reagan MR, Nahas MR, Karp Leaf R, Jain S, Arnason J, Ghobrial IM, Anderson KC, Kufe D, Rosenblatt J, Avigan D. Bone marrow stroma protects myeloma cells from cytotoxic damage via induction of the oncoprotein MUC1. Br J Haematol 2017;176:929-38.
172. Kikuchi J, Kuroda Y, Koyama D, Osada N, Izumi T, Yasui H, Kawase T, Ichinohe T, Furukawa Y. Myeloma cells are activated in bone marrow microenvironment by the CD180/MD-1 complex which senses lipopolysaccharide. Cancer Res 2018;78:1766-78.
173. Gu C, Peng H, Lu Y, Yang H, Tian Z, Yin G, Zhang W, Lu S, Zhang Y, Yang Y. BTK suppresses myeloma cellular senescence through activating AKT/P27/Rb signaling. Oncotarget 2017;8:56858-67.
174. Yang Y, Shi J, Gu Z, Salama ME, Das S, Wendlandt E, Xu H, Huang J, Tao Y, Hao M, Franqui R, Levasseur D, Janz S, Tricot G, Zhan F. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res 2015;75:594-604.
175. Kobune M, Chiba H, Kato J, Kato K, Nakamura K, Kawano Y, Takada K, Takimoto R, Takayama T, Hamada H, Niitsu Y. Wnt3/RhoA/ROCK signaling pathway is involved in adhesion-mediated drug resistance of multiple myeloma in an autocrine mechanism. Mol Cancer Ther 2007;6:1774-84.
176. Di Marzo L, Desantis V, Solimando AG, Ruggieri S, Annese T, Nico B, Fumarulo R, Vacca A, Frassanito MA. Microenvironment drug resistance in multiple myeloma: emerging new players. Oncotarget 2016;7:60698-711.
177. Caivano A, Laurenzana I, De Luca L, La Rocca F, Simeon V, Trino S, D'Auria F, Traficante A, Maietti M, Izzo T, D'Arena G, Mansueto G, Pietrantuono G, Laurenti L, Musto P, Del Vecchio L. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumour Biol 2015;36:9739-52.
178. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, Azab F, Flores LM, Campigotto F, Weller E, Anderson KC, Scadden DT, Ghobrial IM. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest 2013;123:1542-55.
179. Frassanito MA, Rao L, Moschetta M, Ria R, Di Marzo L, De Luisi A, Racanelli V, Catacchio I, Berardi S, Basile A, Menu E, Ruggieri S, Nico B, Ribatti D, Fumarulo R, Dammacco F, Vanderkerken K, Vacca A. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia 2014;28:904-16.
180. Tsuyama N, Ishikawa H, Abroun S, Liu S, Li FJ, Otsuyama K, Zheng X, Obata M, Taniguchi O, Kawano MM. The regulatory mechanism of IL-6-dependent proliferation of human myeloma cells. Hematology 2003;8:409-11.
181. Yang H, Du X, Xi Y. Effects of survivin on FVADT chemotherapy for refractory multiple myeloma. Exp Ther Med 2016;12:771-6.
182. Wagner V, Hose D, Seckinger A, Weitz L, Meißner T, Reme T, Rossi JF, Goldschmidt H, Krämer A, Klein B, Raab MS. Survivin in multiple myeloma: prognostic and therapeutic implications. Blood 2011;118:137.
183. Desplanques G, Giuliani N, Delsignore R, Rizzoli V, Bataille R, Barille-Nion S. Impact of XIAP protein levels on the survival of myeloma cells. Haematologica 2009;94:87-93.
184. Abe S, Hasegawa M, Yamamoto K, Kurata M, Nakagawa Y, Suzuki K, Takizawa T, Kitagawa M. Rapid induction of IAP family proteins and Smac/DIABLO expression after proapoptotic stimulation with doxorubicin in RPMI 8226 multiple myeloma cells. Exp Mol Pathol 2007;83:405-12.
185. Nakagawa Y, Abe S, Kurata M, Hasegawa M, Yamamoto K, Inoue M, Takemura T, Suzuki K, Kitagawa M. IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes. Am J Hematol 2006;81:824-31.
186. De Veirman K, Van Ginderachter JA, Lub S, De Beule N, Thielemans K, Bautmans I, Oyajobi BO, De Bruyne E, Menu E, Lemaire M, Van Riet I, Vanderkerken K, Van Valckenborgh E. Multiple myeloma induces Mcl-1 expression and survival of myeloid-derived suppressor cells. Oncotarget 2015;6:10532-47.
187. Le Gouill S, Podar K, Amiot M, Hideshima T, Chauhan D, Ishitsuka K, Kumar S, Raje N, Richardson PG, Harousseau JL, Anderson KC. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood 2004;104:2886-92.
188. Merz M, Jauch A, Hielscher T, Bochtler T, Schonland SO, Seckinger A, Hose D, Bertsch U, Neben K, Raab MS, Hillengass J, Salwender H, Blau IW, Lindemann HW, Schmidt-Wolf IGH, Scheid C, Haenel M, Weisel KC, Goldschmidt H. Prognostic significance of cytogenetic heterogeneity in patients with newly diagnosed multiple myeloma. Blood Adv 2018;2:1-9.
189. Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, Bauer M, Stein C, Deshpande S, Wardell C, Buzder T, Molnar G, Zangari M, van Rhee F, Thanendrarajan S, Schinke C, Epstein J, Davies FE, Walker BA, Meissner T, Barlogie B, Morgan GJ, Weinhold N. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun 2017;8:268.
190. Hebraud B, Leleu X, Lauwers-Cances V, Roussel M, Caillot D, Marit G, Karlin L, Hulin C, Gentil C, Guilhot F, Garderet L, Lamy T, Brechignac S, Pegourie B, Jaubert J, Dib M, Stoppa AM, Sebban C, Fohrer C, Fontan J, Fruchart C, Macro M, Orsini-Piocelle F, Lepeu G, Sohn C, Corre J, Facon T, Moreau P, Attal M, Avet-Loiseau H. Deletion of the 1p32 region is a major independent prognostic factor in young patients with myeloma: the IFM experience on 1195 patients. Leukemia 2014;28:675-9.
191. Gertz MA, Lacy MQ, Dispenzieri A, Greipp PR, Litzow MR, Henderson KJ, Van Wier SA, Ahmann GJ, Fonseca R. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood 2005;106:2837-40.
192. Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau JL, Bataille R; Intergroupe Francophone du Myélome. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001;98:3082-6.
193. Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, Dawson KJ, Iorio F, Nik-Zainal S, Bignell GR, Hinton JW, Li Y, Tubio JM, McLaren S, S OM, Butler AP, Teague JW, Mudie L, Anderson E, Rashid N, Tai YT, Shammas MA, Sperling AS, Fulciniti M, Richardson PG, Parmigiani G, Magrangeas F, Minvielle S, Moreau P, Attal M, Facon T, Futreal PA, Anderson KC, Campbell PJ, Munshi NC. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014;5:2997.
194. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, Baker A, Bergsagel PL, Bernstein BE, Drier Y, Fonseca R, Gabriel SB, Hofmeister CC, Jagannath S, Jakubowiak AJ, Krishnan A, Levy J, Liefeld T, Lonial S, Mahan S, Mfuko B, Monti S, Perkins LM, Onofrio R, Pugh TJ, Rajkumar SV, Ramos AH, Siegel DS, Sivachenko A, Stewart AK, Trudel S, Vij R, Voet D, Winckler W, Zimmerman T, Carpten J, Trent J, Hahn WC, Garraway LA, Meyerson M, Lander ES, Getz G, Golub TR. Initial genome sequencing and analysis of multiple myeloma. Nature 2011;471:467-72.
195. Kortuem KM, Braggio E, Bruins L, Barrio S, Shi CS, Zhu YX, Tibes R, Viswanatha D, Votruba P, Ahmann G, Fonseca R, Jedlowski P, Schlam I, Kumar S, Bergsagel PL, Stewart AK. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J 2016;6:e397.
196. Johnson DC, Weinhold N, Mitchell JS, Chen B, Kaiser M, Begum DB, Hillengass J, Bertsch U, Gregory WA, Cairns D, Jackson GH, Forsti A, Nickel J, Hoffmann P, Noethen MM, Stephens OW, Barlogie B, Davis FE, Hemminki K, Goldschmidt H, Houlston RS, Morgan GJ. Genome-wide association study identifies variation at 6q25.1 associated with survival in multiple myeloma. Nat Commun 2016;7:10290.
197. Shah V, Boyd KD, Houlston RS, Kaiser MF. Constitutional mutation in CDKN2A is associated with long term survivorship in multiple myeloma: a case report. BMC Cancer 2017;17:718.
198. Basmaci C, Pehlivan M, Tomatir A, Sever T, Okan V, Yilmaz M, Oguzkan-Balci S, Pehlivan S. Effects of TNFalpha, NOS3, MDR1 gene polymorphisms on clinical parameters, prognosis and survival of multiple myeloma cases. Asian Pac J Cancer Prev 2016;17:1009-14.
199. Corre J, Munshi N, Avet-Loiseau H. Genetics of multiple myeloma: another heterogeneity level? Blood 2015;125:1870-6.
200. Andrulis M, Lehners N, Capper D, Penzel R, Heining C, Huellein J, Zenz T, von Deimling A, Schirmacher P, Ho AD, Goldschmidt H, Neben K, Raab MS. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov 2013;3:862-9.
201. Lu S, Yang J, Chen Z, Gong S, Zhou H, Xu X, Wang J. Different mutants of PSMB5 confer varying bortezomib resistance in T lymphoblastic lymphoma/leukemia cells derived from the Jurkat cell line. Exp Hematol 2009;37:831-7.
202. Balsas P, Galan-Malo P, Marzo I, Naval J. Bortezomib resistance in a myeloma cell line is associated to PSMbeta5 overexpression and polyploidy. Leuk Res 2012;36:212-8.
203. Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat 2015;18:18-35.
204. Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, Munker R, Volinia S, Boccadoro M, Garzon R, Palumbo A, Aqeilan RI, Croce CM. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A 2008;105:12885-90.
205. Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014;9:1511-20.
206. Abedin SM, Boddy CS, Munshi HG. BET inhibitors in the treatment of hematologic malignancies: current insights and future prospects. Onco Targets Ther 2016;9:5943-53.
208. Sive JI, Feber A, Smith D, Quinn J, Beck S, Yong K. Global hypomethylation in myeloma is associated with poor prognosis. Br J Haematol 2016;172:473-5.
209. Zhang W, Wang YE, Zhang Y, Leleu X, Reagan M, Zhang Y, Mishima Y, Glavey S, Manier S, Sacco A, Jiang B, Roccaro AM, Ghobrial IM. Global epigenetic regulation of microRNAs in multiple myeloma. PLoS One 2014;9:e110973.
210. Bi C, Chung TH, Huang G, Zhou J, Yan J, Ahmann GJ, Fonseca R, Chng WJ. Genome-wide pharmacologic unmasking identifies tumor suppressive microRNAs in multiple myeloma. Oncotarget 2015;6:26508-18.
211. Dupere-Richer D, Licht JD. Epigenetic regulatory mutations and epigenetic therapy for multiple myeloma. Curr Opin Hematol 2017;24:336-44.
212. Pawlyn C, Kaiser MF, Heuck C, Melchor L, Wardell CP, Murison A, Chavan SS, Johnson DC, Begum DB, Dahir NM, Proszek PZ, Cairns DA, Boyle EM, Jones JR, Cook G, Drayson MT, Owen RG, Gregory WM, Jackson GH, Barlogie B, Davies FE, Walker BA, Morgan GJ. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin Cancer Res 2016;22:5783-94.
213. Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA, Giralt S, Mateos MV, Leleu X, Anderson KC. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia 2018;32:252-62.
214. Sonneveld P, Broijl A. Treatment of relapsed and refractory multiple myeloma. Haematologica 2016;101:396-406.
215. Cornell RF, Kassim AA. Evolving paradigms in the treatment of relapsed/refractory multiple myeloma: increased options and increased complexity. Bone Marrow Transplant 2016;51:479-91.
216. Dimopoulos M, Alegre A, Stadtmauer EA, Goldschmidt H, Zonder JA, de Castro CM, Masliak Z, Reece D, Olesnyckyj M, Yu Z, Weber DM. The efficacy and safety of lenalidomide plus dexamethasone in relapsed and/or refractory multiple myeloma patients with impaired renal function. Cancer 2010;116:3807-14.
217. Dimopoulos M, Siegel DS, Lonial S, Qi J, Hajek R, Facon T, Rosinol L, Williams C, Blacklock H, Goldschmidt H, Hungria V, Spencer A, Palumbo A, Graef T, Eid JE, Houp J, Sun L, Vuocolo S, Anderson KC. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol 2013;14:1129-40.
218. Laubach J, Garderet L, Mahindra A, Gahrton G, Caers J, Sezer O, Voorhees P, Leleu X, Johnsen HE, Streetly M, Jurczyszyn A, Ludwig H, Mellqvist UH, Chng WJ, Pilarski L, Einsele H, Hou J, Turesson I, Zamagni E, Chim CS, Mazumder A, Westin J, Lu J, Reiman T, Kristinsson S, Joshua D, Roussel M, O'Gorman P, Terpos E, McCarthy P, Dimopoulos M, Moreau P, Orlowski RZ, Miguel JS, Anderson KC, Palumbo A, Kumar S, Rajkumar V, Durie B, Richardson PG. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30:1005-17.
219. Matsui W, Borrello I, Mitsiades C. Autologous stem cell transplantation and multiple myeloma cancer stem cells. Biol Blood Marrow Transplant 2012;18:S27-32.
220. Byrne M, Salmasinia D, Leather H, Cogle CR, Davis A, Hsu JW, Wiggins L, Chang MN, An Q, Wingard JR, Moreb JS. Tandem autologous stem cell transplantation for multiple myeloma patients based on response to their first transplant-a prospective phase II study. Clin Med Insights Oncol 2014;8:101-5.
221. Cowan AJ, Stevenson PA, Libby EN, Becker PS, Coffey DG, Green DJ, Hyun TS, Fromm JR, Gopal AK, Holmberg LA. Circulating plasma cells at the time of collection of autologous PBSC for transplant in multiple myeloma patients is a negative prognostic factor even in the age of post-transplant maintenance therapy. Biol Blood Marrow Transplant 2018; doi: 10.1016/j.bbmt.2018.02.017. doi: 10.1016/j.bbmt.2018.02.017.
222. Lee SE, Lim JY, Ryu DB, Kim TW, Jeon YW, Yoon JH, Cho BS, Eom KS, Kim YJ, Kim HJ, Lee S, Cho SG, Kim DW, Lee JW, Min WS, Min CK. Circulating CD3(+)CD4(+)CD161(+) cells are associated with early complications after autologous stem cell transplantation in multiple myeloma. Biomed Res Int 2018;2018:5097325.
223. Pulte ED, Dmytrijuk A, Nie L, Goldberg KB, McKee AE, Farrell AT, Pazdur R. FDA approval summary: lenalidomide as maintenance therapy after autologous stem cell transplant in newly diagnosed multiple myeloma. Oncologist 2018; doi: 10.1634/theoncologist.2017-0440. doi: 10.1634/theoncologist.2017-0440.
224. Micallef IN, Stiff PJ, Nademanee AP, Maziarz RT, Horwitz ME, Stadtmauer EA, Kaufman JL, McCarty JM, Vargo R, Cheverton PD, Struijs M, Bolwell B, DiPersio JF. Plerixafor plus granulocyte colony-stimulating factor for patients with non-Hodgkin lymphoma and multiple myeloma: long-term follow-up report. Biol Blood Marrow Transplant ; doi: 10.1016/j.bbmt.2018.01.039.
225. Gossi U, Jeker B, Mansouri Taleghani B, Bacher U, Novak U, Betticher D, Egger T, Zander T, Pabst T. Prolonged survival after second autologous transplantation and lenalidomide maintenance for salvage treatment of myeloma patients at first relapse after prior autograft. Hematol Oncol 2018;36:436-44.
226. Sonneveld P, Marie JP, Huisman C, Vekhoff A, Schoester M, Faussat AM, van Kapel J, Groenewegen A, Charnick S, Zittoun R, Lowenberg B. Reversal of multidrug resistance by SDZ PSC 833, combined with VAD (vincristine, doxorubicin, dexamethasone) in refractory multiple myeloma. A phase I study. Leukemia 1996;10:1741-50.
227. Dalton WS, Crowley JJ, Salmon SS, Grogan TM, Laufman LR, Weiss GR, Bonnet JD. A phase III randomized study of oral verapamil as a chemosensitizer to reverse drug resistance in patients with refractory myeloma. A Southwest Oncology Group study. Cancer 1995;75:815-20.
228. Friedenberg WR, Rue M, Blood EA, Dalton WS, Shustik C, Larson RA, Sonneveld P, Greipp PR. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): a trial of the Eastern Cooperative Oncology Group. Cancer 2006;106:830-8.
229. Gulla A, Di Martino MT, Gallo Cantafio ME, Morelli E, Amodio N, Botta C, Pitari MR, Lio SG, Britti D, Stamato MA, Hideshima T, Munshi NC, Anderson KC, Tagliaferri P, Tassone P. A 13 mer LNA-i-miR-221 inhibitor restores drug sensitivity in melphalan-refractory multiple myeloma cells. Clin Cancer Res 2016;22:1222-33.
230. Jones RJ, Gu D, Bjorklund CC, Kuiatse I, Remaley AT, Bashir T, Vreys V, Orlowski RZ. The novel anticancer agent JNJ-26854165 induces cell death through inhibition of cholesterol transport and degradation of ABCA1. J Pharmacol Exp Ther 2013;346:381-92.
231. Mark E, Issa MC. Withaferin A induces cell death and differentiation in multiple myeloma cancer stem cells. Med Chem Commun 2016;8:10.
232. Issa ME, Wijeratne EMK, Gunatilaka AAL, Cuendet M. Withanolide D exhibits similar cytostatic effect in drug-resistant and drug-sensitive multiple myeloma cells. Front Pharmacol 2017;8:610.
233. Matsui W, Wang Q, Brennan S, Meeker A, Tressler R, Harley C, Huff CA, Smith BD, Jones R. The novel telomerase inhibitor GRN163L targets drug-resistant cancer stem cells in multiple myeloma. Mol Cancer Ther 2007;6 Suppl 11:A97.
234. Richardson PG, Bensinger WI, Huff CA, Costello CL, Lendvai N, Berdeja JG, Anderson LD, Siegel DS, Lebovic D, Jagannath S, Laubach JP, Stockerl-Goldstein KE, Kwei L, Clow F, Elias L, Salman Z, Graef T, Bilotti E, Vij R. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: phase 2 trial results. Br J Haematol 2018;180:821-30.
235. Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene 2012;31:4257-65.
236. Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai K, Herait P, Kahatt C, Lokiec F, Salles G, Facon T, Palumbo A, Cunningham D, Zucca E, Thieblemont C. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol 2016;3:e196-204.
237. Drew AE, Moradei O, Jacques SL, Rioux N, Boriack-Sjodin AP, Allain C, Scott MP, Jin L, Raimondi A, Handler JL, Ott HM, Kruger RG, McCabe MT, Sneeringer C, Riera T, Shapiro G, Waters NJ, Mitchell LH, Duncan KW, Moyer MP, Copeland RA, Smith J, Chesworth R, Ribich SA. Identification of a CARM1 inhibitor with potent in vitro and in vivo activity in preclinical models of multiple myeloma. Sci Rep 2017;7:17993.
238. Sonneveld P, Suciu S, Weijermans P, Beksac M, Neuwirtova R, Solbu G, Lokhorst H, van der Lelie J, Dohner H, Gerhartz H, Segeren CM, Willemze R, Lowenberg B; European Organization for Research and Treatment of Cancer (EORTC); Leukaemia Cooperative Group (LCG); Dutch Haemato-Oncology Cooperative Study Group (HOVON). Cyclosporin A combined with vincristine, doxorubicin and dexamethasone (VAD) compared with VAD alone in patients with advanced refractory multiple myeloma: an EORTC-HOVON randomized phase III study (06914). Br J Haematol 2001;115:895-902.
239. Dimopoulos K, Sogaard Helbo A, Fibiger Munch-Petersen H, Sjo L, Christensen J, Sommer Kristensen L, Asmar F, Hermansen NEU, O'Connel C, Gimsing P, Liang G, Gronbaek K. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol Oncol 2018;12:180-95.
240. Alzrigat M, Parraga AA, Majumder MM, Ma A, Jin J, Osterborg A, Nahi H, Nilsson K, Heckman CA, Oberg F, Kalushkova A, Jernberg-Wiklund H. The polycomb group protein BMI-1 inhibitor PTC-209 is a potent anti-myeloma agent alone or in combination with epigenetic inhibitors targeting EZH2 and the BET bromodomains. Oncotarget 2017;8:103731-43.
241. Jakubowiak A, Offidani M, Pegourie B, De La Rubia J, Garderet L, Laribi K, Bosi A, Marasca R, Laubach J, Mohrbacher A, Carella AM, Singhal AK, Tsao LC, Lynch M, Bleickardt E, Jou YM, Robbins M, Palumbo A. Randomized phase 2 study: elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016;127:2833-40.
242. Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, Belch A, Krishnan A, Vescio RA, Mateos MV, Mazumder A, Orlowski RZ, Sutherland HJ, Blade J, Scott EC, Oriol A, Berdeja J, Gharibo M, Stevens DA, LeBlanc R, Sebag M, Callander N, Jakubowiak A, White D, de la Rubia J, Richardson PG, Lisby S, Feng H, Uhlar CM, Khan I, Ahmadi T, Voorhees PM. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387:1551-60.
243. Schonfeld K, Zuber C, Pinkas J, Hader T, Bernoster K, Uherek C. Indatuximab ravtansine (BT062) combination treatment in multiple myeloma: pre-clinical studies. J Hematol Oncol 2017;10:13.
244. Karadimitris A, Chaidos A, Caputo V, Goudevenou K, Ponnusamy K, Xiao X. Myeloma propagating cells, drug resistance and relapse. Stem Cells 2015;33:3205-11.
245. Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, Penco S, Pisano C, Carminati P, Tortoreto M, Zunino F, Vlodavsky I, Sanderson RD, Yang Y. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res 2011;17:1382-93.
246. Vasuthasawat A, Yoo EM, Trinh KR, Lichtenstein A, Timmerman JM, Morrison SL. Targeted immunotherapy using anti-CD138-interferon alpha fusion proteins and bortezomib results in synergistic protection against multiple myeloma. MAbs 2016;8:1386-97.
247. Kulkarni U, Karsten CM, Kohler T, Hammerschmidt S, Bommert K, Tiburzy B, Meng L, Thieme L, Recke A, Ludwig RJ, Pollok K, Kalies K, Bogen B, Boettcher M, Kamradt T, Hauser AE, Langer C, Huber-Lang M, Finkelman FD, Kohl J, Wong DM, Manz RA. IL-10 mediates plasmacytosis-associated immunodeficiency by inhibiting complement-mediated neutrophil migration. J Allergy Clin Immunol 2016;137:1487-97.e6.
248. Muthu Raja KR, Rihova L, Zahradova L, Klincova M, Penka M, Hajek R. Increased T regulatory cells are associated with adverse clinical features and predict progression in multiple myeloma. PLoS One 2012;7:e47077.
249. Comeau JM, Kelly K, Jean GW. The role of elotuzumab in the treatment of relapsed or refractory multiple myeloma. Am J Health Syst Pharm 2018;75:55-66.
250. Frerichs KA, Nagy NA, Lindenbergh PL, Bosman P, Marin Soto J, Broekmans M, Groen RWJ, Themeli M, Nieuwenhuis L, Stege C, Nijhof IS, Mutis T, Zweegman S, Lokhorst HM, van de Donk N. CD38-targeting antibodies in multiple myeloma: mechanisms of action and clinical experience. Expert Rev Clin Immunol 2018;14:197-206.
251. Qin H, Wei G, Sakamaki I, Dong Z, Cheng WA, Smith DL, Wen F, Sun H, Kim K, Cha S, Bover L, Neelapu SS, Kwak LW. Novel BAFF-receptor antibody to natively folded recombinant protein eliminates drug-resistant human B-cell malignancies in vivo. Clin Cancer Res 2018;24:1114-23.
252. Yang C, Xiong F, Wang J, Dou J, Chen J, Chen D, Zhang Y, Luo S, Gu N. Anti-ABCG2 monoclonal antibody in combination with paclitaxel nanoparticles against cancer stem-like cell activity in multiple myeloma. Nanomedicine (Lond) 2014;9:45-60.
253. Study of Natalizumab in Relapsed/Refractory Multiple Myeloma. In: U.S. National Library of Medicine; 2008. ClinicalTrials.gov Identifier: NCT00675428. .
254. Podar K, Zimmerhackl A, Fulciniti M, Tonon G, Hainz U, Tai YT, Vallet S, Halama N, Jager D, Olson DL, Sattler M, Chauhan D, Anderson KC. The selective adhesion molecule inhibitor natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: therapeutic implications. Br J Haematol 2011;155:438-48.
255. Sherbenou DW, Mark TM, Forsberg P. Monoclonal antibodies in multiple myeloma: a new wave of the future. Clin Lymphoma Myeloma Leuk 2017;17:545-54.
257. Fernandez-Poma SM, Salas-Benito D, Lozano T, Casares N, Riezu-Boj JI, Mancheno U, Elizalde E, Alignani D, Zubeldia N, Otano I, Conde E, Sarobe P, Lasarte JJ, Hervas-Stubbs S. Expansion of tumor-infiltrating CD8(+) T cells expressing PD-1 improves the efficacy of adoptive t-cell therapy. Cancer Res 2017;77:3672-84.
258. Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, He X, Devine SM, Zhang X, Caligiuri MA, Hofmeister CC, Yu J. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014;28:917-27.
259. Larocca A, Palumbo A. Optimizing treatment for elderly patients with newly diagnosed multiple myeloma: a personalized approach. J Clin Oncol 2016; doi: 10.1200/JCO.2016.68.6113. doi: 10.1200/JCO.2016.68.6113.
260. Hari PN, Zhang MJ, Roy V, Perez WS, Bashey A, To LB, Elfenbein G, Freytes CO, Gale RP, Gibson J, Kyle RA, Lazarus HM, McCarthy PL, Milone GA, Pavlovsky S, Reece DE, Schiller G, Vela-Ojeda J, Weisdorf D, Vesole D. Is the international staging system superior to the Durie-Salmon staging system? A comparison in multiple myeloma patients undergoing autologous transplant. Leukemia 2009;23:1528-34.
261. Kastritis E, Terpos E, Roussou M, Gavriatopoulou M, Migkou M, Eleutherakis-Papaiakovou E, Fotiou D, Ziogas D, Panagiotidis I, Kafantari E, Giannouli S, Zomas A, Konstantopoulos K, Dimopoulos MA. Evaluation of the revised international staging system in an independent cohort of unselected patients with multiple myeloma. Haematologica 2017;102:593-9.
262. Agarwal MB. Multiple myeloma: treatment is getting individualized. Indian J Hematol Blood Transfus 2016;32:3-9.
263. van Rhee F, Szymonifka J, Anaissie E, Nair B, Waheed S, Alsayed Y, Petty N, Shaughnessy JD Jr, Hoering A, Crowley J, Barlogie B. Total therapy 3 for multiple myeloma: prognostic implications of cumulative dosing and premature discontinuation of VTD maintenance components, bortezomib, thalidomide, and dexamethasone, relevant to all phases of therapy. Blood 2010;116:1220-7.
264. Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, Kumar S, Rajkumar SV, Carpten JD, Barrett M, Fonseca R, Stewart AK, Bergsagel PL. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120:1067-76.
265. Sikic BI. Modulation of multidrug resistance: a paradigm for translational clinical research. Oncology (Williston Park) 1999;13:183-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Nass J, Efferth T. Drug targets and resistance mechanisms in multiple myeloma. Cancer Drug Resist 2018;1:87-117. http://dx.doi.org/10.20517/cdr.2018.04
AMA Style
Nass J, Efferth T. Drug targets and resistance mechanisms in multiple myeloma. Cancer Drug Resistance. 2018; 1(2): 87-117. http://dx.doi.org/10.20517/cdr.2018.04
Chicago/Turabian Style
Nass, Janine, Thomas Efferth. 2018. "Drug targets and resistance mechanisms in multiple myeloma" Cancer Drug Resistance. 1, no.2: 87-117. http://dx.doi.org/10.20517/cdr.2018.04
ACS Style
Nass, J.; Efferth T. Drug targets and resistance mechanisms in multiple myeloma. Cancer Drug Resist. 2018, 1, 87-117. http://dx.doi.org/10.20517/cdr.2018.04
About This Article
Copyright

Data & Comments
Data


 Cite This Article 24 clicks
Cite This Article 24 clicks

 Like This Article 26
likes
Like This Article 26
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.