
How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance?


Abstract
P-glycoprotein (ABCB1), multidrug resistance protein-1 (ABCC1) and breast cancer resistance protein (ABCG2) belong to the ATP-binding cassette (ABC) superfamily of proteins that play an important physiological role in protection of the body from toxic xenobiotics and endogenous metabolites. Beyond this, these transporters determine the toxicity profile of many drugs, and confer multidrug resistance (MDR) in cancer cells associated with a poor treatment outcome of cancer patients. It has long been hypothesized that inhibition of ABC drug efflux transporters will increase drug accumulation and thereby overcome MDR, but until now no approved inhibitor of these transporters is available in the clinic. In this review we present molecular strategies to overcome this type of drug resistance and discuss for each of these strategies their promising value or indicate underlying reasons for their limited success.
Keywords
Introduction
One of the most challenging obstacles in cancer treatment is multidrug resistance (MDR). Despite having achieved lower mortality rates due to recent advances in cancer therapy, the long term survival rate remains poor, primarily due to chemotherapy resistance[1-3]. Resistance can be caused by numerous mechanisms in cancer cells, such as activation of drug-metabolizing enzymes (e.g., cytochrome P450), activation of DNA repair mechanisms, disruptions in apoptotic signaling pathways, reduced drug influx and increased activity of drug efflux pumps[4-6]. Specifically, several members of the ATP-binding cassette (ABC) superfamily mediate the efflux of multiple chemotherapeutic drugs and so contribute to MDR. The ABC superfamily represents one of the largest protein families in biology, including 49 ABC genes identified in the human genome[7,8]. This superfamily is subdivided into seven distinct groups (ABCA through ABCG) and currently at least 15 ABC transporters have been implicated to confer resistance to clinically active drugs, notably P-glycoprotein (P-gp, ABCB1), multidrug resistance protein-1 (MRP1, ABCC1) and breast cancer resistance protein (BCRP, ABCG2)[9].
Structurally, each ABC transporter is composed of at least one hydrophobic membrane-spanning domain (MSD) consisting of 6 α-helices and 1 hydrophilic nucleotide binding domain (NBD). All ABC proteins share a highly conserved region in the ATP binding domain, including the Walker A and Walker B sequences, the ABC signature motif, the H loop and the Q loop[8]. Depending on the clustering of the MSD and the NBD in the transporters, these are grouped into full transporters (e.g., ABCB1 and ABCC1, consisting of 2 MSDs and 2 NBDs), half transporters (e.g., ABCG2, containing only 1 MSD and 1 NBD) and non-transporters, which possess two NBDs, without the MSDs[10]. The subfamily ABCE belongs to the non-transporters, whose proteins play a role in the assembly of the pre-initiation complex and RNAse inhibition[11,12]. ABCF subfamily members also encode non-transporters, they are involved in ribosome assembly and protein translation[13,14].
ABC transporters are present in all living species, in prokaryotes they function as drug importers or exporters, in eukaryotes solely as drugs exporters[15]. By coupling ATP binding and hydrolysis these transporters are able to extrude substrates out of cells or into cellular compartments like cytoplasmic vesicles and lysosomes[8]. This superfamily transports a wide range of substrates, including lipids, bile salts, sugars, amino acids, steroids, peptides, nucleotides, endogenous metabolites, ions and toxins, including antibiotics and chemotherapeutic drugs. Collectively, ABC transporters fulfill both physiological and pharmacological functions impacting drug transport, tumor growth modulation and regulatory pathways (e.g., apoptosis and complement-mediated cytotoxicity[16-18] and immune cell-regulatory pathways[19]). This review focuses on current developments in drug discovery to circumvent MDR induced by P-gp, MRP1 and BCRP in cancer cells.
P-glycoprotein
P-glycoprotein (or ABCB1 or MDR1) was originally identified as the first mammalian ABC multidrug transporter in 1976, when Juliano and Ling[20] observed that Chinese hamster ovary cells displayed pleiotropic cross-resistance to a wide range of amphiphilic drugs. This transporter was named P-gp and is also known as ABCB1 or MDR1.
P-gp is encoded by a single polypeptide chain with 2 homologous NBD and 2 homologous transmembrane domains (TMD)[21,22]. The 2 homologous TMD are joined by a linker region of about 60 amino acids[23]. Each TMD contains 6 transmembrane (TM) α-helices and 1 NBD [Figure 1].
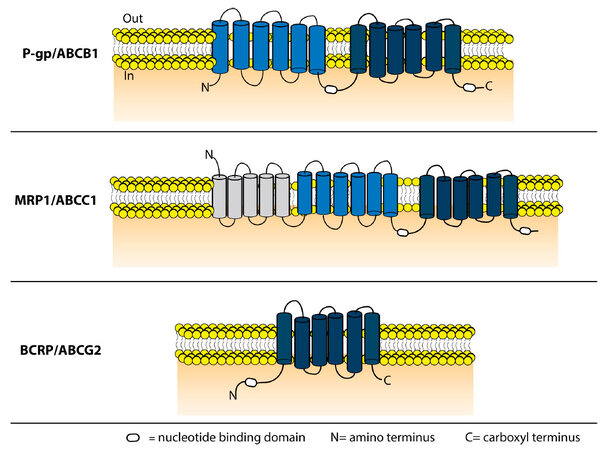
Figure 1. Schematic representation of the domain structure of P-gp, MRP1 and BCRP. P-gp has 2 TMD consisting of 6 α-helices each and 2 ATP bindings sites. MRP1 also contains 2 ATP-binding regions and 2 TMD containing 6 α-helices and it is extended by 1 additional TMD consisting of 5 α-helices and 1 amino terminus loop (L0). The half transporter ABCG2 contains 1 TMD of 6 α-helices and 1 ATP-binding domain. The ATP-binding site of this transporter is found on the amino-terminal side (N) in contrast to P-gp and MRP1. P-gp: P-glycoprotein; MRP1: multidrug resistance protein-1; BCRP: breast cancer resistance protein; TMD: transmembrane domain
Aller et al.[24] reported the X-ray structure of mouse P-gp, which shares 87% sequence identity with the human P-gp. The structure revealed that the inward facing conformation is formed by 2 bundles of 6 TMs arranged as TMs 1-3, 6, 10, 11 and TMs 4, 5, 7-9, 12. This conformation results in a large internal cavity, which is open to the cytoplasm and the inner leaflet. There are 2 portals (formed by TMs 4/6 and 10/12) that allow hydrophobic molecules to enter the cavity directly from the inner leaflet. The drug binding pocket generally contains hydrophobic and aromatic residues and is large enough to accommodate at least two substrate molecules simultaneously[25]. In fact, this was supported by the X-ray structure reported by Aller et al.[24]. Expanding on these observations, Pajeva and Wiese[26] compared the inward facing homology model of human P-gp based on the earlier mentioned mouse structure with the outward facing homology model of human P-gp based on Sav1866 structure. It appeared that residues in TM4 and TM10 face the cavity in the inward facing conformation are completely buried in the outward facing form, while the ligands remain bound to the same residues of TM6 and TM12 in both conformations. This implies that each of the portals has different roles: TM4 and TM10 as “portal keepers” (preventing substrates that entered to escape back to the inner leaflet) and TM6 and TM12 as “portal carriers” (these are mainly responsible for substrate interactions). Together, this model suggests that ligands remain bound to the same residues during the transition from the inward to the outward facing conformation.
The expression pattern of P-gp indicates that its main function is the protection of the body from toxic substances and xenobiotics by excreting them into bile, urine, feces and avoiding their entry into fetus, brain and testes. To this end, P-gp is expressed on the canalicular surface of hepatocytes in the liver, the apical surface of epithelial cells of proximal tubules in the kidneys, columnar epithelial cells of the intestine, epithelial cells of placenta, and the luminal surface of capillary endothelial cells in brain and testes[27,28][Table 1]. Next to this important physiological function, pharmacologically P-gp constitutes a considerable obstacle to the delivery of various clinically used drugs to their targets, such as anti-cancer drugs, anti-epileptics, immunosuppressive agents, cardiac glycosides, cholesterol-lowering statins, human immunodeficiency virus (HIV) protease inhibitors, anti-hypertensives, calcium channel blockers, anti-histamines and antibiotics[15]. From knockout mice studies it became clear that P-gp plays a major role in determining the drug ADME (absorption, distribution, metabolism, excretion) profile[29,30]. These P-gp knock-out mice showed increased absorption and decreased elimination of drugs, which gave rise to severe toxicity. In addition, P-gp works synergistically with the major phase I drug metabolizing enzyme P450 (CYP3A4), as they share a remarkable similarity between their substrates and regulation, to decrease the oral drug bioavailability.
Characterization of P-gp, MRP1 and BCRP
| ABC transporter | Localization | High expression | Low expression | Substrates |
|---|---|---|---|---|
| P-gp (ABCB1) | Apical | Kidney, adrenal glands, liver, pancreas, intestine, lung, BBB, placenta | Prostate, skin, heart/skeletal ovaries | Neutral, positively charged hydrophobic substrates |
| MRP1 (ABCC1) | Basolateral (except in placenta and BBB) | Kidney, lung, testis, skeletal/cardiac muscle, placenta | Liver, intestine, brain | Anionic substrates in the form of anionic glutathione, glucuronate or sulfate conjugates |
| BCRP (ABCG2) | Apical | Placenta, breast, BBB, liver, intestine | Kidney, lung, ovaries, testis, pancreas | + or − charged, hydrophobic/hydrophilic conjugated/unconjugated |
High expression levels of P-gp have been identified in various solid tumor malignancies, e.g., kidney, colon, liver, ovarian, breast and sarcomas[4]. P-gp overexpression has been reported to be an independent negative prognostic factor in clinical outcome. In high grade osteosarcoma, increased levels of P-gp correlated with a > 3-fold increased risk of adverse events and shorter event-free survival[31]. P-gp RNA levels increased after lung perfusion with doxorubicin of patients with unresectable sarcoma pulmonary metastases[32]. Furthermore, Gregorcyk et al.[33] reported that P-gp positive breast cancer patients are at significantly higher risk for relapse. In hematological malignancies, P-gp mRNA expression and protein function are increased after chemotherapy treatment. Expression of P-gp in myeloid blasts is correlated with treatment failure and shorter survival in adult acute myeloid leukemia (AML) patients[34,35]. However, childhood acute lymphoblastic leukemia (ALL) results have shown contradictory results, some found no association between high P-gp expression and prognosis[36,37], while other groups showed prognostic impact for P-gp in ALL[38,39]. These differences can at least partially be attributed to different treatment strategies and methods used to detect P-gp[40].
Multi-drug associated protein 1
A decade after the discovery of P-gp, the first member of the C subfamily, MRP1 (systematic name, ABCC1) was discovered[41]. MRP1 is widely expressed in normal tissues, with relatively high levels in the kidney, lung, testis, heart and placenta, while there is a low expression in colon, brain, and small intestine and peripheral blood mononuclear cells[42,43][Table 1]. Notably, in the above-mentioned tissues, MRP1 expression can vary between different cell types of the same tissue[44]. The highest expression is found in cells with specialized barrier function, e.g., the choroid cells in the blood cerebrospinal fluid barrier[45], or cells with a high proliferative status, e.g., reactive type II pneumocytes in the alveoli of the lung[46]. In contrast to P-gp and BCRP, MRP1 are mainly expressed at the basolateral membrane of polarized cells, with the exception of brain capillary endothelial cells. Consequently, it has been suggested that MRP1 functions to protect cell types from xeno- and endobiotics and pumping them into the interstitial space of the body, instead of their expulsion into bile, urine or gut[47]. In addition, studies with mrp1 (-/-) mice revealed other physiologic functions of MRP1 than transporters. For example, MRP1 plays a role in immunological responses by its involvement in leukotriene extrusion[48]. Furthermore, being a transporter of glutathione (GSH) and glutathione-conjugates, MRP1 plays a crucial role in the elimination of toxic substances formed during oxidative stress[49,50].
Despite the fact that both P-gp and MRP1 can confer resistance to a wide range of drugs, P-gp prefers neutral, or positively charged, hydrophobic compounds, while MRP1 binds and transports anionic substrates, mainly in the form of anionic glutathione, glucuronate or sulfate conjugates[51][Table 1]. Unmodified drugs can also be co-transported with free glutathione[50]. Some examples of MRP1 substrates are: various anticancer drugs, antibiotics, pesticides, HIV protease inhibitors, peptides, heavy metals, glutathione conjugates, glucuronide conjugates and sulfate conjugates[15].
The currently accepted MRP1 topology consists of the following domain arrangement: TMD0-L0-TMD1-NBD1-TMD2-NBD2 [Figure 1]. Compared to P-gp, this transporter is extended by an additional TMD0 containing 5 TM helices and 1 amino terminus loop (L0) consisting of 32 amino acids, which links TMD0 to TMD1. The exact function of the TMD0 still needs to be fully elucidated. It was previously thought that TMD0 had no role in trafficking to the plasma membrane or efflux function, but this view has been revisited by several mechanistic and cell biological reports[52]. It has been shown that TMD0 is important for the processing and trafficking of human MRP1[53]. Additionally, by mutation studies, Yang et al.[54] reported that the amino terminus is required for proper MRP1 function and structure. It has also been suggested by Chen et al.[55] that the amino terminus forms a U-shaped structure which might serve as a gate to regulate the substrate transport activity of MRP1. Finally, TMD0 and L0 seem to contribute to MRP1 homo-dimerization, but whether the functional form of MRP1 is a homodimer or a monomer needs to be determined[56].
Depending on expression levels, MRP1 can confer resistance to a variety of antineoplastic drugs, including vinca alkaloids, anthracyclines, epipodophyllotoxins, saquinavir, methotrexate, mitoxantrone, camptothecins, paclitaxel, glucuronide, doxorubicin, epirubicin, and tyrosine kinase inhibitors such as imatinib[57,58]. High MRP1 expression levels have been identified in different cancer types, e.g., lung cancer, breast cancer, prostate cancer, gastrointestinal carcinoma, melanoma, neuroblastoma, ovarian and hematological malignancies[44,59] (AML, ALL and chronic lymphoblastic leukemia). In these results, it has been difficult to define statistically significant correlations between high MRP1 expression in the indicated tumors and acquired resistance or prognosis. This was largely due to the fact that tumor samples contained a variable number of MRP1 expressing normal cells, cells, while other drug efflux transporters might contribute as well[44]. However, a prospective study demonstrated that MRP1 is an independent prognostic indicator of outcome in neuroblastoma patients[60]. In addition, a high expression of MRP1 was associated with shorter tumor-free survival (TFS) and overall survival (OS) in non-small cell lung cancer (NSCLC)[61]. Similarly, in breast cancer MRP1 expression was related to shorter relapse-free survival (RFS) and OS[62].
Breast cancer resistance protein
ABCG2, also known as BCRP, ABCP or MXR, is expressed in a variety of normal tissues, including epithelial cells of the gastrointestinal tract and liver, placental syncytiotrophoblast cells, prostate epithelium, kidney cortical tubules, endothelial cells of the blood-brain barrier (BBB) and hematopoietic stem cells[10,63][Table 1]. Like P-gp, ABCG2 is expressed on the apical surface of epithelial cells and since it is expressed in tissues with secretory and barrier function, this suggests that its function is similar to P-gp, which is protecting the body from toxic compounds. ABCG2 mRNA expression was higher compared to other efflux transporter in the human small intestine, which is a rate-limiting barrier to oral drug absorption[64]. The important impact of ABCG2 on the intestinal absorption was further supported by several knockout mice research, which have indicated that ABCG2 affects the pharmacological and toxicological behavior of many drugs[65-67]. Also, polymorphisms might influence the role of ABCG2 in intestinal absorption. For example, ABCG2 c.421C> polymorphism renders BCRP more than 75% less active than its wild-type variant. Since ABCG2 c.421C>A is a common polymorphism, present in more than 30% of the Asian and 15% of European population, decreased drug absorption due to BCRP polymorphisms deserves clinical attention[68]. Additionally, Kruijtzer et al.[69] reported that ABCG2 affects the bioavailability and systemic concentration of topotecan in patients. Inhibition of ABCG2 by elacridar (GF120918), a dual inhibitor of P-gp and ABCG2, increased the bioavailability of topotecan from 40% to 97% after oral administration and reduced the biliary and renal excretion of topotecan after intravenous administration. It has also been noted that the polymorphic variant 421C>A of ABCG2 can alter the pharmacokinetics of several drugs, including diflomotecan, teriflunomide, sulfasalazine, gefitinib, imatinib, and many statins[63]. Furthermore, it has been suggested that ABCG2 plays a role in the maintenance of human pluripotent stem cells in an undifferentiated state and their protection from toxic xenobiotics and hypoxia[10].
BCRP exhibits broad substrate specificity and can transport either positively or negatively charged drugs, hydrophobic or hydrophilic and conjugated or unconjugated substrates[70]. Examples are a wide range of anticancer drugs, sulfate and glucuronide conjugates of xenobiotics, tyrosine kinase inhibitors (TKIs), statins, fluorescent dyes and flavonoids[10,63]. In vitro assays showed that ABCG2 might have more than two binding sites, since ABCG2 inhibitors displayed different inhibition profiles, depending on the tested substrate[71-73]. For instance, the inhibitor Ko143 inhibited the transport of all the substrates tested by Giri et al.[72], which suggested its binding to a region that allosterically inhibits the transport of these substrates. However, the ABCG2 inhibitors elacridar, nelfinavir and Pluronic P85 seemed to bind to a different region, which inhibited the transport of the nucleoside analogs abacavir and zidovudine, but showed no or a partial effect on the transport of prazosin and imatinib. Muenster et al.[73] showed that two different substrates, such as topotecan and albendazole sulfoxide, could simultaneously be transported without hindering each other’s transport, which also suggest multiple binding sites in ABCG2. Furthermore, using homology modeling a complete homodimer ABCG2 model was created based on the crystal structure of the bacterial multidrug exporter Sav1866, which also suggested a multiple binding site[74]. ABCG2 is a half transporter consisting of 6 putative transmembrane segments and 1 NBD [Figure 1]. The simplest functional form of ABCG2 is a homodimer, which is bridged by disulfide bridge bonds[75]. Other research has suggested higher order of homo-oligomer on plasma membranes, which could also regulate ABCG2 function by dynamic association and dissociation of ABCG2 monomers[76]. Accordingly, it is currently unknown whether a homodimer or a homo-oligomer is the dominant functional unit of ABCG2 in the plasma membrane[77,78].
Clinically, high expression levels of ABCG2 have been found in many solid tumors, especially adenocarcinomas of the digestive tract, endometrium, lung and melanoma[77]. In addition, Damiani et al.[79] found that ABCG2 was overexpressed in 33% of AML patients and that this feature was significantly associated with shorter disease-free survival and higher risk of relapse. ABCG2 expression is also higher in B-lineage than in T-lineage ALL, while adult ALL patients have a higher expression than infant ALL patients[80,81]. Contradictory results have been published on the correlation between ABCG2 expression and prognosis of solid tumors and on the correlation between ABCG2 expression and prognosis of hematological malignancies[10,39,82]. Overall, even though BCRP activity contributes to MDR and drug pharmacokinetics in cancer, the exact mechanisms of action and interaction remain to be elucidated.
ABC transporter inhibition
Since overexpression of some of the ABC transporters was correlated with a poor chemotherapeutic response and prognosis of patients with specific cancer types, inhibition of ABC transporters appears a logical approach to circumvent MDR and improve patient’s outcome[82,83]. However, other than in a preclinical setting where ABC transporters could reverse MDR, this promise has still not been fulfilled in clinical practice. Inhibition of ABC transporters has been evaluated with rationally designed or natural inhibitors, including competitive and non-competitive inhibitors. Competitive inhibitors exert their function by tightly binding and blocking the substrate binding pockets. Non-competitive inhibitors exert their function by binding to a non-substrate binding site thereby inhibiting the ATPase activity or modulating transporters’ function allosterically[15,83-85]. Another strategy to circumvent MDR that will be discussed further is the application of small interfering RNA (siRNA)[86-88] and microRNA (miRNA)[89,90] inoculation to down-regulate ABC transporter expression. Currently, approximately 30 siRNA candidates are being studied in clinical trial, yet unfortunately their high cost, poor stability in vivo, delivery challenges and off-target effects still remain a challenge for treatment[89]. A third approach relates to the rational design of new chemotherapeutics which are non-substrates for ABC transporters. TKIs are small molecules developed to inhibit the uncontrolled activity of various tyrosine kinases (TK) involved in cancer. Many of them can also act as substrates or non-substrates to alter efflux mechanisms mediated by ABC transporters[91]. TKIs have been approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) since 2001 (such as imatinib, gefitinib and erlotinib and more recently ceritinib, lenvatinib) while several others are under investigation in clinical trials and can potentially be used to inhibit transporter-mediated efflux, as discussed below[92-96]. The main focus of this section of the review will be to discuss modulators which are lead compounds for clinical application.
Overcoming P-gp resistance
P-gp specific inhibition is particularly challenging due to its large binding pocket with low substrate specificity which enables P-gp to interact with over 200 different known substrates[9,97]. Nevertheless, the discovery of new agents to inhibit P-gp is still ongoing, building on encouraging in vitro studies and individual clinical trials such as the Southwest Oncology Group (SWOG 9126) study that showed a strong benefit of the use of the P-gp inhibitor cyclosporine A (CSA) in patients with relapsed or refractory AML[98]. To date, three generations of P-gp inhibitors can be distinguished. Both, the first-generation agents (including verapamil, tamoxifen, cyclosporine A, quinidine and dexverapamil), and the second-generation agents [including VX-710 (biricodar), GF120918 (elacridar) and PSC833 (valspodar)] ultimately failed to show improvement in overall drug efficacy in multiple randomized clinical trials due to poor potency and increased toxicity, respectively[99]. Consequently, the third-generation agents were developed featuring a high transporter affinity and a low pharmacokinetic interaction, including XR-9576 (Tariquidar), LY335979 (Zosuquidar) and R101933 (Laniquidar), which will be discussed below.
Inhibition of P-gp
Agents from all the three generations have been tested in clinical trials. Even though some of the early trials showed benefit from P-gp inhibition, results of most large randomized trials have been disappointing[100,101]. As an illustration, a phase I study of tariquidar in combination with vinorelbine showed that tariquidar is a potent P-gp inhibitor, with no noteworthy side effects and pharmacokinetic interactions[102]. Similar findings were observed for zosuquidar. Phase I research with zosuquidar showed that the drug could be administered safely to patients with AML or solid tumors[103,104]. However, a randomized placebo-controlled double-blind trial in 449 patients with newly diagnosed AML or high-risk myelodysplastic syndrome, showed no improvement in OS, or complete remission (CR) rates of drug treatment combined with zosuquidar[105].
The main reason for the initial failures in early clinical trials which started within 10 years after the discovery of P-gp-mediated MDR, was the lack of full understanding of P-gp properties and lack of knowledge on other ABC transporters. A clear example of this is tariquidar, which initially was considered as a specific P-gp inhibitor, but later results showed inhibitory effects on BCRP and MRP1 leading to the synthesis of more potent tariquidar derivatives[106,107]. Mechanistic studies also showed that tariquidar can act, in a dose-dependent manner, both as a substrate or as a transport inhibitor[106,108,109]. Furthermore, recent 3-dimensional quantitative structure-activity relationship (QSAR) studies of anthranilamide derivatives of tariquidar suggest that a hydrophobic domain is needed for increased P-gp specific inhibitory function[26]. Some tariquidar analogs with sulfonamide groups are P-gp specific, nevertheless tariquidar was toxic to healthy cells[107]. Since its discovery, tariquidar was described as non-competitive and non-transported inhibitor of P-gp[110], while Bankstahl et al.[111] have shown that tariquidar is concentration-dependently transported by P-gp. Recent transepithelial drug transport assays using radioactive tariquidar in human and mouse cell lines indicate that tariquidar functions as a high-affinity P-gp substrate rather than a non-competitive inhibitor[112].
The insufficient knowledge of the P-gp transporter led to the design of clinical trials with inadequate inhibitors and patient selection. Agents such as verapamil belonging to the first-generation inhibitors were low in potency, and since dose escalation was limited due to toxicity, ineffective dosages were used[113,114]. Other clinical trials failed due to their choice of compounds that interfered with the physiological role of P-gp and sister proteins. For instance, the dose of the anticancer agents used in combination with the second-generation P-gp inhibitor PSC 833 had to be reduced to prevent toxicities of the anticancer agent because PSC 833 caused a decreased drug clearance via inhibition of its metabolism by cytochrome P450[112]. Regarding patient selection, initial clinical trials enrolled patients irrespective of their P-gp status, whereas rationally patient selection should be based on adequate transporter(s) expression and/or function in tumor specimen and whether the targeted transporter is the dominant mechanism of drug resistance[91]. Moreover, early clinical trial designs did not take into account the role of P-gp polymorphisms. Polymorphic variants of the transporters influence their expression, function and localization and thereby might affect the pharmacokinetics of their substrates[115]. Inclusion of patients with such polymorphic variants might confuse treatment outcome assessments, since these patients might not develop significant MDR via efflux pumps, but face bone marrow toxicity in combination therapy with the inhibitor.
Novel molecules that can successfully interact with P-gp-specific NBDs have been identified through in silico and computational studies (compound 19, 29, 45)[116,117]. The aim of such approach is to find molecules that can interfere with the energy harvesting system of a transporter, avoiding its non-specific drug binding domains. Nanayakkara et al.[118] identified through computational studies (compound 29, 34, 45) which, when co-administrated with vinblastine and paclitaxel decreased viability, survival and motility of prostate cancer and other resistant cell lines. Furthermore compound 34 and 45 were found to be P-gp specific while inhibitor 45 also had affinity for ABCG2[117]. These techniques may be a promising approach in the search for P-gp specific NBD inhibitors[119].
Evading P-gp (nanoparticle drug delivery systems)
Focus has now shifted towards downregulating the transcription of P-gp with siRNA[87,120,121] or microRNA[122,123] using nanoparticle (NP) drug delivery systems[124]. Hyaluronic acid based nanoparticles can effectively target CD44+ ovarian cancer cells and downregulate P-gp and increase intracellular concentration of paclitaxel. High CD44+ expression in ovarian cancer cells is related to metastasis, therefore this cell-specific approach has the potential to improve the cell sensitivity in ovarian cancer patients with poor prognosis[120,121]. Encapsulation drugs in liposomal nanoparticles show promising results in the clinical setting. Doxorubicin HCl liposomal injection (Doxil) was approved for the treatment of AIDS-related Kaposi's sarcoma (USA in 1995 and Japan in 2007), and for the treatment of ovarian cancer (USA in 1999 and Japan in 2009) after failure of prior systemic chemotherapy or intolerance or after failure of platinum-based chemotherapy[125]. In addition, in 2007 the FDA approved Doxil for the treatment of multiple myeloma in combination with bortezomib. Another candidate is the nanoparticle albumin-bound paclitaxel (nab-paclitaxel, Abraxane), which in 2011 has been approved in 42 countries for the treatment of metastatic breast cancer after failure of combination chemotherapy for metastatic disease or relapse within six months after adjuvant chemotherapy[126]. Additionally, encapsulation of doxorubicin in hydrophobic polymeric micelles cause indirect inhibition of P-gp efflux and enhance intracellular drug concentration without affecting P-gp expression[127]. The BBB is also another target of investigation with NP drug delivery. A recent study analyzed the permeability kinetics of siRNA NPs in a BBB monolayer model using human brain endothelial cells. It was concluded that NP delivery can inhibit P-gp expression by half and increase rhodamine 123 permeability by a third[87]. siRNA-loaded NP carriers could be a promising approach to circumvent the human BBB in order to facilitate drug penetration in the central nervous system. Unfortunately, side effects such as skin and hypersensitivity reactions and neurotoxicity remain common limiting factor[128,129].
The role of microRNAs in P-gp modulation
MicroRNAs are post-transcriptional regulators composed of noncoding RNAs, which can regulate gene expression and have been implicated in drug resistance in many cancers[122,123]. MicroRNA can bind to the 3’ untranslated region (UTR) of RNA with high precision, causing RNA degradation and inhibition of gene expression[130]. Several studies report that miRNAs can regulate MDR by modulating the expression of P-gp. For example, it has been shown that miR-451 and miR-27a upregulation was P-gp dependent in cell lines representing ovarian cancer, leukemia and hepatocellular carcinoma[112,114,115]. In contrast, deregulation rather than upregulation of certain miRNAs, such as miR27a and miR 331-5p, can also cause drug resistance reversal and decreased P-gp expression, as seen in doxorubicin resistant leukemia cell lines K562 and HL60[131-133]. Furthermore one study found a possible prognostic value for miR-296, whose expression can distinguish long-term survivors from short-term survivors, while its downregulation sensitized cells to known P-gp and BCRP substrates[134].
MiRNA mediated P-gp inhibition and drug resistance reversal seem to be dose-dependent, as seen with colorectal adenocarcinoma and breast cancer cell lines[135,136]. For example, Bao et al.[136] reported that P-gp expression could be reduced with miR-298 and reversed doxorubicin resistance in breast cancer cells in a dose-dependent fashion. In vivo studies also report that miR-873 elicits a dose-dependent response, measured by tumor growth inhibition and cisplatin sensitization in previously resistant cell lines[122]. Other miRNAs with BCRP-modulating capacity include miR-122[137], miR-381, miR-495[138], miR-223[139], miR-9[140], and miR-873[122][Table 2].
Novel approaches to P-gp inhibition
| Type | Name | Remarks | Cancer type | References |
|---|---|---|---|---|
| P-gp inhibitors | Tariquidar, zosuquidar Laniquidar cyclosporine tariquidar derivatives | Zosuquidar shows robust P-gp inhibition; tariquidar + vinrelbine displayed potent P-gp inhibition without affecting survival rates; hydrophobic domains in tariquidar derivatives increases inhibition | AML/high-risk MDS | [26,98-112] |
| Computational studies/in silico drug screening | Compound 19, -29, -34, -45 | High NBD specificity; compounds 34 and 45 are P-gp specific | Prostate cancer and various MDR cell lines | [116-119] |
| Nanoparticle drug delivery systems | Encapsulated drugs: doxorubicin, paclitaxel, rhodamine 123 | Increases drug concentration and indirectly inhibit P-gp | Ovarian cancer, Kaposi sarcoma | [87,121,124-129] |
| siRNA | siRNA delivered by nanoparticles | Hyaluronic acid based nanoparticles for CD44+ cell, reduces P-gp mRNA expression by half in BBB model with NP delivery and over 85% in Caco-2 cells without NP delivery, in dose-dependent manner | Ovarian cancer, BBB cell-based model, Caco-2 cell line | [87,120,121,135] |
| MicroRNAs | miR-27a, -451, -873, -381, -101, -229, -495, -9, -296, -298 | Inhibits P-gp expression through post-transcriptional modulation; increases drug sensitivity in previously resistant cells | Breast, ovarian cancer, hepatocellular, colorectal carcinoma, leukemia | [122,123,130-134,138,140] |
| Non-substrates | Microtubule stabilizing/de-stabilizing agents, topoisomerase inhibitors | Taxanes such as ixabepilone is FDA approved beta microtubulin mutations lead to resistance | Breast cancer | [141-149] |
| Monoclonal antibodies | MRK-16, 17 | Currently undergoing phase I clinical trials | Leukemia | [9,172-175] |
| TKIs | Alectinib, ibrutinib, neratinib, AG-1393, EKI-785, vandetinib | Sensitized primary resistant cells and cell lines to chemotherapeutics by inhibiting P-gp ATPase activity | AML/ALL, several others | [91-96,181-183] |
Unfortunately, full understanding of miRNA post-transcriptional modulation is still insufficient and concerns with off target effects on top of which RNA degradation still remains an unressolved issue[89].
Bypassing P-gp: none substrates
Several approaches have been investigated to evade P-gp mediated MDR with novel drugs: (1) decreasing their molecular affinity for P-gp and (2) increasing the affinity to drug targets. This has been challenged for microtubule stabilizing agents (e.g., epothilones, second- and third-generation taxanes), microtubule destabilizing agents (e.g., cryptophycins, halichondrins, hemiasterlins and STX140), inhibitors of topoisomerase I (e.g., lipophilic camptothecins, homocamptothecins and dibenzonaphthyridinones) and inhibitors of topoisomerase II (e.g., lipophilic anthracyclines). Many of these agents have already undergone wide preclinical and clinical investigation, and are comprehensively reviewed[141].
The epothilones including epothilone B (patupilone) and its analogs BMS-310705, sagopilone and ixabepilone, and epothilone D and its derivative KOS-1584, are poor substrates of P-gp. They show a diverse susceptibility profile to P-gp mediated resistance, with ixabepilone the most and sagopilone the less affected by P-gp overexpression[142,143]. Currently, ixabepilone is FDA approved for the treatment of metastatic breast cancer and has been tested in phase III clinical trials in combination with capecitabine[144-146].
TPI 287, a novel microtubule destabilizing drug of the taxane family designed to circumvent MDR, was recently tested in a small group phase I study of children with neuroblastoma and medulloblastoma. TPI 287 was well tolerated in half of patients with neuroblastoma who completed one cycle of treatment with TPI 287 alone showed a stable disease although the study was not designed to evaluate treatment response[147]. In the case of epothilones, there are a number of active clinical trials to test its efficacy in a range of advanced solid tumors such as breast cancer, ovary, head and neck, esophagus, lung, gastrointestinal tract cancers and brain tumors (clinical trial reference: NCT00030173, NCT0035516 and NCT00450866). Being a P-gp substrate, ixabepilone might exert its antitumor activity preferentially in tumors where the main resistance mechanism is βIII-tubulin or/and BCRP dependent[148,149]. The exact working mechanism of these novel agents is still unclear. However, bypassing a resistance mechanism such as P-gp-mediated MDR does not imply that patients are not at risk of developing other mechanisms of drug resistance which will lead to intrinsic or acquired tumor refractoriness. For epothilones, expression of some MRPs, such as MRP7, and β-tubulin mutations has been related to the development of tumor drug resistance to these agents[150,151].
Using P-gp to kill cancer cells
Instead of inhibition of the P-gp function or evading its efflux to overcome P-gp-mediated MDR, an alternative approach is to actually exploit P-gp expression to specifically kill MDR cells. This strategy makes use of the phenomenon of collateral sensitivity (CS). which refers to the fact that some agents show preferential activity toward MDR cancer cells relative to their non-MDR parental cells, as was previously noticed for verapamil, cytosine arabinoside and gemcitabine in MDR small cell lung cancer cells lines[152,153]. Different CS agents have been synthesized from lead compounds such as NSC73306 (1,1-Isatin-4-(4′-methoxyphenyl)-3-thiosemicarbazone) and the flavonoid desmosdumotin B[154,155]. It has been hypothesized that CS agents exert their effect by the generation of reactive oxygen species. This strategy is still experimental. Of further interest, within a class of isatins drugs, particularly N-alkylisatins were found not to be susceptible to P-gp mediated efflux, destabilize microtubule formation and induce apoptosis in P-gp overexpressing tumor cells[156].
Another approach to bypass P-gp is to use drugs that show a higher sensitivity to P-gp expressing cells compared to P-gp negative cells. Bergman et al.[153,157] showed that both small cell lung cancer and NSCLC cells with an overexpression of either P-gp or MRP1 were more sensitive to the deoxynucleoside analogs cytarabine and gemcitabine. This was related to an increased activity of the activating enzyme deoxycytidine kinase (dCK) for these nucleoside analogs, leading to an increased accumulation of the active metabolites, ara-CTP and dFdCTP, respectively. It was argued that in P-gp cells steroids are effluxed efficiently decreasing their intracellular concentration. Since steroids regulate dCK, this may lead to an increased enzyme activity[158]. The increased sensitivity was observed in cells with acquired resistance to P-gp substrates, such as etoposide, doxorubicin, colchicine, but also in cells transfected with the gene for P-gp or MRP1[153,157].
Increase oral bioavailability by increasing gut epithelial uptake
A strategy that can benefit of P-gp inhibition is the increase of the oral bioavailability of drugs, particularly when a drug is a P-gp substrate and is efficiently being effluxed back to the gut leading to a low oral bioavailability. The oral administration of drugs has several advantages over intravenous formulation[159]. First of all, patients prefer oral chemotherapy over intravenous due to the ease of administration, avoiding the discomfort of injection and the related risk of infection[159,160]. Secondly, oral medication is cost saving for hospital and health insurance, since there is no need for hospitalization. Finally, oral administration of drugs enables metronomic therapy with the advantages of chronic exposure of the drugs to its targets and reducing dose-related toxicities, such as the optimizing of anti-angiogenic efficacy of chemotherapeutic agents including paclitaxel and docetaxel[161,162]. Wild-type mice showed limited oral bioavailability of paclitaxel compared P-gp knockout mice[163]. Following this, multiple preclinical mice results have shown that inhibition of P-gp using inhibitors such as cyclosporine, PSC 833 or elacridar improved the oral treatment of paclitaxel in mice[164-166]. Therefore, clinical trials were started to explore the effect of the oral administration of paclitaxel in combination with cyclosporine. Three phase ІІ trials have tested this combination in patients with NSCLC, advanced gastric cancer and breast cancer. These studies have shown promising results, but long-term oral use of cyclosporine might be complicated by immunosuppressive effects. Therefore, other inhibitors might replace cyclosporine such as elacridar (GF120918) or HM30181, respectively[167,168]. Being a P-gp substrate in addition to the encouraging results of oral paclitaxel, preclinical studies were also initiated with another taxane, namely docetaxel[169]. Different inhibitors were used such as OC144-093 (a P-gp-inhibitor), cyclosporine (a dual inhibitor of P-gp and CYP3A4) and ritonavir (CYP3A4 inhibitor), which have shown increased oral bioavailability of docetaxel[170]. In line with this observation, which indicates that CYP3A4 is the major determinant of the low oral bioavailability of docetaxel, it seems rational that future trials will continue with oral docetaxel in combination with ritonavir[167,170].
Other studies have shown that P-gp restricts the oral bioavailability of the topoisomerase II inhibitor, etoposide, and that P-gp inhibition enhances its oral bioavailability[171]. The appropriate inhibitor and associated dosages (of both drug and inhibitor) still need to be determined to improve the therapeutic outcome with minimal toxicities, since P-gp influences the excretion of drugs in addition to its role in protecting of vital organs such as the brain, the fetus and the testes. Furthermore, attention needs to be paid to specific polymorphisms of P-gp that might impact the drug oral bioavailability.
Monoclonal antibodies against P-gp
Already in the 1986 Hamada and Tsuruo[172] developed two monoclonal antibodies against human P-gp in adriamycin-resistant myelogenous leukemia cells (MRK-16 and MRK-17). Initially, these antibodies were intended to study the membrane changes in MDR cells, but later research demonstrated that MRK-16 was able to inhibit P-gp mediated efflux of vincristine and actinomycin D in vivo[172,173]. The addition of a P-gp inhibitor such as cyclosporine A to MRK-16 treatment, further sensitized MDR myelogenous leukemia cells to vincristine and doxorubicin[174,175]. This innovative approach with MRK-16 is currently being investigated again in phase I clinical trials[9].
Overcoming MRP1 resistance
MRP1 was identified as a drug transporter 15 years after P-gp, and therefore its role in MDR in human tumors has not been fully understood and fewer MRP1-specific inhibitors with sufficient potency and efficacy have been identified[176]. Several novel strategies that have been applied to P-gp are being tried on MRP1 overexpressing cells are listed on [Table 3]. Nanoparticle drug delivery systems have been also applied successfully to inhibit MRP1 mediated resistance[177,178]. For example, Wang et al.[177] used redox-responsive polymeric micelles with a disulfide bond that serves as a linker for delivery of paclitaxel as chemotherapeutic, and indomethacin, as chemosensitizer. Also, siRNA delivery in porous silicon nanoparticles has shown to effectively downregulate MRP1 mRNA[178] yet more studies are needed, especially in vivo.
Novel approaches to MRP1 inhibition
| Type | Name | Remarks | Cancer type | References |
|---|---|---|---|---|
| MRP1 inhibitors | Tariquidar, dofequidar cyclosporine A, biricodar, CBT-1 and derivatives of triazolonaphthalimide | Triazolonaphtalimide is anti-proliferative effect in MRP1 overexpressing lung cancer cells | Lung cancer, others | [184-189] |
| Nanoparticle drug delivery systems | Agents delivered: paclitaxel, indomethacin and siRNA | Redox-responsive polymeric micelles with a disulfide bond for delivery of paclitaxel and indomethacin. Porous silicon nanoparticles protect siRNA from degradation | Breast cancer, glioblastoma | [177,178] |
| TKIs | Alectinib, ibrutinib, neratinib, AG-1393, EKI-785, vandetinib | Sensitized primary resistant cells and cell lines to chemotherapeutics by inhibiting P-gp ATPase activity | AML/ALL, several others | [179-183] |
As with P-gp, numerous TKIs have been also identified to modulate the efflux of MRP1. Cediranib (AZD217) is an orally administered potent small-molecule TKI that inhibits the vascular endothelial growth factor (VEGF) family of receptors (VEGFR 1-3), platelet-derived growth factor receptor (PDGFR) and a stem cell factor receptor, c-Kit[179]. A study by Tao et al.[180] showed that cediranib inhibited the ATPase activity of P-gp in a dose - dependent manner and it also reversed both P-gp and MRP1 mediated MDR, suggesting that cediranib acts as dual inhibitor. Another TKI, vandetanib (ZD6474, Zactima), an inhibitor of VEGFR, epidermal growth factor receptor (EGFR) and rearranged during transfection (RET) tyrosine kinases[181], stimulated P-gp ATPase activity in a dose-dependent manner and reversed MDR in cancer cells by directly inhibiting the function of P-gp, MRP1 and ABCG2[181,182]. Other TKIs such as AG-1393 and EKI-785, have also been found to interact and inhibit P-gp activity[183].
Among the three generations of P-gp inhibitors, there are inhibitors that also inhibit MRP1 and/or ABCG2. Examples of such inhibitors are cyclosporine A[26,184-187], biricodar (VX-710), dofequidar (MS-209), tariquidar and CBT-1. The novel triazolonaphtalimide derivatives have been reported to achieve an anti-proliferative effect in chemoresistant lung cancer cell lines via MRP1 downregulation without affecting P-gp function[188]. Some of these preclinical studies reached phase III clinical trials, for instance for dofequidar[189] and CBT-1 (NCT00437749). However, inadequate trial design and poor drug specificity might hamper proper assessments of the full potential of these agents. Further investigation is warranted to assess the effect of these inhibitors on each targeted ABC transporter in clinical trials, to guide selection of the right population of patients to enhance the efficacy and minimize the toxicity.
The scarcity of specific MRP1 inhibitors reflects the fact that there is still no clarity about the benefit of P-gp inhibition in the clinical practice, which retards research efforts to develop MRP1 inhibitors for clinical use[190]. However, the calcium channel blocker verapamil and its derivative NMeOHI2, in addition to the several non-steroidal anti-inflammatory drugs (e.g., indomethacin), have shown collateral sensitivity toward MRP1 overexpressing cells[191,192]. These agents were proposed to trigger apoptosis in cells overexpressing MRP1 via intracellular GSH depletion. Furthermore, through high-throughput screening and chemical modification studies, the tricyclic isoxazole derivatives were identified to exhibit high potency and specificity to MRP1 inhibition. Nevertheless, further studies are needed to address MRP1 inhibition specificity, toxicity and effectivity both in vivo and in vitro.
Overcoming ABCG2 resistance
Ahmed-Belkacem et al.[193] divided ABCG2 inhibitors in four groups: (1) ABCG2-specific inhibitors; (2) broad-spectrum inhibitors, including inhibitors that inhibit P-gp and/or MRP1; (3) TKIs; and (4) naturally occurring flavonoids and derivatives.
ABCG2 inhibitors
The mycotoxin fumitremorgin C (FTC) was identified as the first ABCG2 specific inhibitor, but its neurotoxic effect hampered its further clinical development[194,195]. This lead compound was the basis of a new non-toxic tetracyclic analogue of FTC, Ko143, which is a highly potent and specific inhibitor of ABCG2[195]. Ko143 increased topotecan plasma concentration 4-6-fold in Mdr1a/1b-/- mice. Pick et al.[196] identified a new class of specific ABCG2 inhibitors which were structurally related to tariquidar, but did not show potency toward P-gp and MRP1. In their research they presented evidence for 3 different binding sites on ABCG2, one occupied by the ABCG2 substrate pheophorbide A (Pheo A), another by the TKI imatinib and a third by the two compounds that specifically inhibit ABCG2. Imatinib showed features of an allosteric modulator by increasing the inhibitory effect of the inhibitor compounds. Another ABCG2 inhibitor lead compound, PZ-39, was reported by Peng et al.[197]. Interestingly, this compound had a dual mode of action; it could effectively inhibit ABCG2 function and markedly reduced the half-life of ABCG2 protein from approximately 54 to 5 h by accelerating its protein degradation via endocytosis and trafficking to lysosomes. The ABCG2 inhibitor specificity of these compounds was confirmed with respect to P-gp and MRP1, but not for other ABC transporters.
Transcriptional down regulation using siRNA or miRNA are also areas of interest in ABCG2 inhibition. One study shows that combination of siRNA and PI3K/Akt signaling inhibition can reverse chemoresistance in mitoxantrone-resistant BCRP-overexpressing cells lines[198][Table 4]. Similarly, nanoparticle drug delivery using polyethyleneimine as a carrier for siRNA has been reported to down-regulate ABCG2 and decrease the mitoxantrone IC(50) values by approximately 14-fold[86]. Other efforts to deliver ABCG2 and P-gp inhibitors with nanoparticles show complete reversal of resistance in ABCG2 positive MDR cells[199].
Novel approaches to ABCG2 inhibition
| Type | Name | Remarks | Cancer type | References |
|---|---|---|---|---|
| ABCG2 inhibitors | Tariquidar derivatives: PZ-39, FTC derivatives: Ko-143 | Ko-143 is highly specific,PZ-39 reduces half-life of ABCG2 | Colon carcinoma, resistant cell lines | [194-197] |
| TKIs | Imatinib, gefitinib | By competitive inhibition, TKIs act as substrates at low concentrations and inhibitor-like properties at higher concentrations | CML, NSCLC | [94,208-214] |
| Nanoparticle drug delivery systems | Encapsulated agents:mitoxantrone siRNA | Complete reversal of resistance in ABCG2 positive MDR cells | Resistant cell lines | [86,198,199] |
| siRNA | siRNA delivered by nanoparticles | Polyethyleneimine as a carrier for siRNA inhibits ABCG2 and sensitizes cells to mitoxantrone 14-fold | Resistant cell lines | [86,198] |
| MicroRNAs | miR-328, -hsa-miR-328, miR -519, -520h, miR-212, -181a, miR 487a and -302 | Use of microRNA has revealed a more complex role of ABCG2 in drug resistance related signaling pathways: SIRT1/CREB/ABCG2 and Wnt/β-catenin-ABCG2 pathway | Breast, ovarian cancer, other cell lines | [200-207] |
There are several miRNA that have been identified in a wide range of cancer cells, as potential inhibitors of ABCG2 expression (such as miRNA-328, -hsa-miR-328, -519, -520h, -212, -181a, 487a and miR-302[200-205]. The use of miRNA to interfere with transporter transcription confirmed the role of ABCG2 in drug resistance related signaling pathways. For example, it was seen that miR-132 enhanced cisplatin resistance in gastric cancer cells by targeting SIRT1 which regulates ABCG2 expression by promoting the de-acetylation of the transcription factor CREB (SIRT1/CREB/ABCG2 pathway). Moreover, Kaplan-Meier survival analysis showed that low miR-132 is related to longer survival in chemo resistant gastric cancer[206]. Another example is Wnt/β-catenin-ABCG2 pathway, which is modulated by miR-199a/b in colorectal cancer stem cells, and whose upregulation is related to cisplatin resistance[207].
Dual inhibitors
Some of the broad-spectrum inhibitors of P-gp, including elacridar, reserpine, cyclosporine A, tariquidar and PSC 833 also display ABCG2 inhibitory activity[10]. Such dual inhibitors will be effective in cases where both transporters participate in MDR and the cytotoxic drug is a substrate of both transporters. Conceivably, their inhibition will also increase the oral bioavailability of the drug, in addition to its penetration to the tumor site. However, since these two transporters are also expressed in sanctuary sites, caution for potential toxic side effects should be considered. Moreover, this awareness would also hold for cytotoxic drugs with a narrow therapeutic window, as minor elevations of drug exposure might lead to adverse reactions.
TKIs as ABCG2 inhibitor or substrate
Various TKIs are either a substrate, an inhibitor or both, for P-gp, MRP1 and ABCG2[168]. TKIs such as the BCR-ABL kinase inhibitors (imatinib, nilotinib and dasatinib), EGFRs kinase inhibitors (gefitinib, erlotinib, lapatinib) and others, show substrate-like properties at lower concentration and inhibitor-like properties at higher concentration via competitive inhibition of the transporters function[208]. In addition to the concentration-dependent type of interaction, it has been proposed that exposure time to TKIs also plays a role[209]. Short exposure (≤ 24 h) to either gefitinib or vandetanib demonstrated a synergic interaction with SN-38, whereas prolonged exposure (5 days) showed a strong antagonism between gefitinib or vandetanib and SN-38. Gefitinib and EK-785 at a low concentration (0.1 to 1 µmol/L) significantly stimulated the ATPase activity, suggesting that these TKIs are transported substrates of ABCG2 whereas higher concentrations induced strong inhibition of ABCG2[210]. Different types of interactions and corresponding possible effects have been suggested by different research groups, which are demonstrated in a hypothetical model [Figure 2].
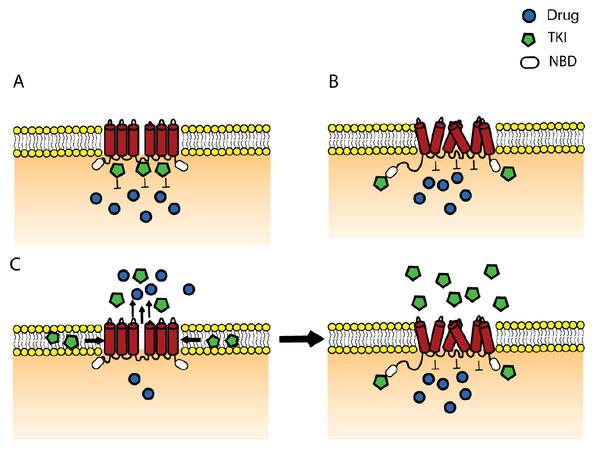
Figure 2. Hypothetical model demonstrating the different possible interactions of TKIs with ABC transporters. (A) The competitive binding of TKI at the drug binding site and hereby preventing the binding of the drug and consequently its efflux; (B) the situation when TKI is not a substrate of ABC transporter, but it binds at the NBD or other site inducing allosteric modulation of the transporter which leads to its inhibition; (C) the case when the TKI behaves as a substrate at low concentration, where the "vacuum cleaner" model is applicable[30]. However, when the concentration increases at a given moment saturation takes place and TKIs penetrate into the cell and bind at the NBD and thereby inhibiting the transporter. ABC: ATP-binding cassette; TKI: tyrosine kinase inhibitor; NBD: nucleotide binding domain
Many combination therapies of TKIs with cytotoxic drugs have been performed (e.g., the combination of gefitinib with 5-fluorouracil, leucovorin, and irinotecan in patients with colorectal cancer, the combination of imatinib and cytarabine in newly diagnosed patients with chronic myeloid leukemia, and the combination of gemcitabine/cisplatin with gefitinib in NSCLC[95,211-214]. TKIs also interact with ABCG2 that can modify ADME-Tox profile of ABCG2 substrates. Gefitinib enhanced the oral bioavailability of irinotecan and topotecan, and increased their apparent bioavailability and decreased systemic clearance in mice[215,216]. Therefore, studies are needed that investigate the exact type of interactions between TKIs and cytotoxic drugs[95,208,209].
Inhibitors from natural sources
ABCG2 inhibitors originating from natural products have been referred to as the fourth-generation inhibitors. Originally, these compounds showed effects in cancer prevention[217], but later on they were found to exert sensitizing effects to chemotherapeutics. Among them are the flavonoids which are polyphenolic compounds found in foods and herbal products. Different flavonoids are potent modulators of ABC transporters: kaempferol, apigenin and myricetin are inhibitors of MRP1[218], kaempferide, quercetin, diosmin and glabridin are inhibitors of P-gp[219] and silymarin, hesperetin and daidzein are inhibitors of ABCG2[220]. The bioflavonoids neochamaejasmin B (NCB)[221] and chamaechromone from the root of Stellera chamaejasme L. are able to inhibit the expression and activity of MRP1. Interestingly, chamaechromone is MRP1 specific, while NCB is a substrate for MRP1 and P-gp. Furthermore NCB and chamaechromone co-administration increased chamaechromone bioavailability substantially[166]. However, further investigation is needed to define optimal dose schedules of flavonoids capable of inducing ABC transporter inhibition[219]. Future studies would also have to take into account flavonoid pharmacokinetics, bioavailability and activity of flavonoid metabolites after ingestion, which would reduce active flavonoid plasma concentrations[222,223].
Conclusion
Efflux transporters of the ABC superfamily including P-gp, MRP1 and ABCG2 can confer MDR in cancer cells. Additionally, overexpression of these transporters in tumor tissues has been associated with poor therapy outcome of cancer patients. Conceptually, inhibition of these ABC transporters will increase drug accumulation and thereby overcome MDR. However, despite accumulating knowledge of ABC transporters and their complex interplay, clinical trials evaluating the effect of ABC transporter inhibitors did not fulfill their promises. In part, this has also been accounted for by poor patient selection criteria.
The design of ABC transporter inhibition research would require several considerations. The choice for a specific inhibitor will increase the likelihood of undesirable side effects when the inhibitor targets multiple ABC transporters, although due to redundancy of ABC transporters, sanctuary sites might be spared from the toxic insults of the cytotoxic drug. When multiple ABC transporters are involved in provoking the MDR phenotype, broad-spectrum inhibitors should be preferred, but as a double-edged sword will also impact physiological functions of these ABC transporters. The evading strategy may serve an alternative for the ABC transporter inhibition dilemma. At this stage, however, the magic bullet that escapes each of the 15 ABC transporters implicated in cancer drug resistance has not been identified and current studies aim to identify those compounds evading the major ABC transporters P-gp, MRP1 and BCRP. Also, MDR evading strategies using encapsulation of drugs have potential downsides such as immune reactions. Lastly, concerning the exploiting strategy, those drugs which cause collateral sensitivity would promote selective targeting of MDR cells, but this concept has not yet been sufficiently explored in animal and clinical studies.
Collectively, each of the MDR reversal strategies has its merits but did not translate in successful clinical application. Future directions should therefore be aimed at combining our current knowledge of clinically relevant ABC transporters in the field of molecular biology (including polymorphic variants), biochemistry, computational biology and sensitive detection and imaging techniques, which in conjunction with state of the art medicinal chemistry can generate new generation inhibitors for future MDR reversal studies. Such an integrated approach may also help to guide personalized therapy in cancer patients to achieve the most optimal treatment outcome.
Declarations
Authors’ contributionsAll authors contributed equally to the paper.
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets 2013;13:326-46.
3. Tiwari AK, Sodani K, Dai CL, Ashby CR Jr, Chen ZS. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr Pharm Biotechnol 2011;12:570-94.
4. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002;2:48-58.
5. Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol 2013;4:28.
6. Cree IA, Charlton P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017;17:1-8.
7. Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 2001;11:1156-66.
8. Vasiliou V, Vasiliou K, Nebert DW. Human ATP-binding cassette (ABC) transporter family. Hum Genomics 2009;3:281-90.
9. Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, Yang DH, Chen ZS. Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 2016;27:14-29.
10. Mo W, Zhang JT. Human ABCG2: structure, function, and its role in multidrug resistance. Int J Biochem Mol Biol 2012;3:1-27.
11. Zhao Z, Fang LL, Johnsen R, Baillie DL. ATP-binding cassette protein E is involved in gene transcription and translation in Caenorhabditis elegans. Biochem Biophys Res Commun 2004;323:104-11.
12. Bisbal C, Martinand C, Silhol M, Lebleu B, Salehzada T. Cloning and characterization of a RNAse L inhibitor. A new component of the interferon-regulated 2-5A pathway. J Biol Chem 1995;270:13308-17.
13. Marton MJ, Vazquez de Aldana CR, Qiu H, Chakraburtty K, Hinnebusch AG. Evidence that GCN1 and GCN20, translational regulators of GCN4, function on elongating ribosomes in activation of eIF2alpha kinase GCN2. Mol Cell Biol 1997;17:4474-89.
14. Tyzack JK, Wang X, Belsham GJ, Proud CG. ABC50 interacts with eukaryotic initiation factor 2 and associates with the ribosome in an ATP-dependent manner. J Biol Chem 2000;275:34131-9.
15. Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2007;9:105-27.
16. Cinci L, Luceri C, Bigagli E, Carboni I, Paccosi S, Parenti A, Guasti D, Coronnello M. Development and characterization of an in vitro model of colorectal adenocarcinoma with MDR phenotype. Cancer Med 2016;5:1279-91.
17. Pallis M, Russell N. P-glycoprotein plays a drug-efflux-independent role in augmenting cell survival in acute myeloblastic leukemia and is associated with modulation of a sphingomyelin-ceramide apoptotic pathway. Blood 2000;95:2897-904.
18. Zhang F, Zhang H, Wang Z, Yu M, Tian R, Ji W, Yang Y, Ruifang N. P-glycoprotein associates with Anxa2 and promotes invasion in multidrug resistant breast cancer cells. Biochem Pharmacol 2014;87:292-302.
19. van de Ven R, Oerlemans R, van der Heijden JW, Scheffer GL, de Gruijl TD, Jansen G, Scheper RJ. ABC drug transporters and immunity: novel therapeutic targets in autoimmunity and cancer. J Leukoc Biol 2009;86:1075-87.
20. Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976;455:152-62.
21. Ferry D, Russell M, Cullen M. P-glycoprotein possesses a 1,4-dihydropyridine-selective drug acceptor site which is alloserically coupled to a vinca-alkaloid-selective binding site. Biochem Biophys Res Commun 1992;188:440-5.
22. Gottesman MM, Ambudkar SV, Xia D. Structure of a multidrug transporter: crystal structures of a mammalian multidrug efflux pump bound to peptide inhibitors may reveal drug binding sites. Nat Biotechnol 2009;27:546-7.
23. Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human "MDR1" gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci U S A 1987;84:3004-8.
24. Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009;323:1718-22.
25. Loo TW, Bartlett MC, Clarke DM. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J Biol Chem 2003;278:39706-10.
26. Pajeva IK, Wiese M. Structure-activity relationships of tariquidar analogs as multidrug resistance modulators. AAPS J 2009;11:435.
27. Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A 1987;84:7735-8.
28. Cordon-Cardo C, O'Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem 1990;38:1277-87.
29. Anwar-Mohamed A, El-Kadi A. P-glycoprotein effects on drugs pharmacokinetics and drug-drug-interactions and their clinical implications. Libyan J Pharm Clin Pharmacol 2012;1:48154.
31. Baldini N, Scotlandi K, Barbanti-Bròdano G, Manara MC, Maurici D, Bacci G, Bertoni F, Picci P, Sottili S, Campanacci M, Serra M. Expression of P-glycoprotein in high-grade osteosarcomas in relation to clinical outcome. N Engl J Med 1995;333:1380-5.
32. Abolhoda A, Wilson AE, Ross H, Danenberg PV, Burt M, Scotto KW. Rapid activation of MDR1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clin Cancer Res 1999;5:3352-6.
33. Gregorcyk S, Kang Y, Brandt D, Kolm P, Singer G, Perry RR. P-glycoprotein expression as a predictor of breast cancer recurrence. Ann Surg Oncol 1996;3:8-14.
34. McLornan DP, McMullin MF, Johnston P, Longley DB. Molecular mechanisms of drug resistance in acute myeloid leukaemia. Expert Opin Drug Metab Toxicol 2007;3:363-77.
35. Xia CQ, Smith PG. Drug efflux transporters and multidrug resistance in acute leukemia: therapeutic impact and novel approaches to mediation. Mol Pharmacol 2012;82:1008.
36. Choi JH, Lim HY, Joo HJ, Kim HS, Yi JW, Kim HC, Cho YK, Kim MW, Lee KB. Expression of multidrug resistance-associated protein1, P-glycoprotein, and thymidylate synthase in gastric cancer patients treated with 5-fluorouracil and doxorubicin-based adjuvant chemotherapy after curative resection. Br J Cancer 2002;86:1578-85.
37. Gao Y, Liao Y, Shen JK, Feng Y, Choy E, Cote G, Harmon D, Mankin HJ, Hornicek FJ, Duan Z. Evaluation of P-glycoprotein (Pgp) expression in human osteosarcoma by high-throughput tissue microarray. J Orthop Res 2016;34:1606-12.
38. Brozek J, Bryl E, Płoszyńska A, Balcerska A, Witkowski JM. P-glycoprotein activity predicts outcome in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2009;31:493-9.
39. Steinbach D, Legrand O. ABC transporters and drug resistance in leukemia: was P-gp nothing but the first head of the Hydra? Leukemia 2007;21:1172.
40. Broxterman HJ, Sonneveld P, Feller N, Ossenkoppele GJ, Währer DC, Eekman CA, Schoester M, Lankelma J, Pinedo HM, Löwenberg B, Schuurhuis GJ. Quality control of multidrug resistance assays in adult acute leukemia: correlation between assays for P-glycoprotein expression and activity. Blood 1996;87:4809-16.
41. Cole S, Bhardwaj G, Gerlach J, Mackie J, Grant C, Almquist K, Stewart A, Kurz E, Duncan A, Deeley R. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992;258:1650.
42. Flens MJ, Zaman GJ, van der Valk P, Izquierdo MA, Schroeijers AB, Scheffer GL, van der Groep P, de Haas M, Meijer CJ, Scheper RJ. Tissue distribution of the multidrug resistance protein. Am J Pathol 1996;148:1237-47.
43. Kourti M, Vavatsi N, Gombakis N, Sidi V, Tzimagiorgis G, Papageorgiou T, Koliouskas D, Athanassiadou F. Expression of multidrug resistance 1 (MDR1), multidrug resistance-related protein 1 (MRP1), lung resistance protein (LRP), and breast cancer resistance protein (BCRP) genes and clinical outcome in childhood acute lymphoblastic leukemia. Int J Hematol 2007;86:166-73.
44. Bakos E, Homolya L. Portrait of multifaceted transporter, the multidrug resistance-associated protein 1 (MRP1/ABCC1). Pflugers Arch 2007;453:621-41.
45. Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, Piwnica-Worms D. Choroid plexus epithelial expression of MDR1 P-glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc Natl Acad Sci U S A 1999;96:3900-5.
46. Bréchot JM, Hurbain I, Fajac A, Daty N, Bernaudin JF. Different pattern of MRP localization in ciliated and basal cells from human bronchial epithelium. J Histochem Cytochem 1998;46:513-7.
47. Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 2005;204:216-37.
48. Wijnholds J, Evers R, van Leusden MR, Mol CA, Zaman GJ, Mayer U, Beijnen JH, van der Valk M, Krimpenfort P, Borst P. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat Med 1997;3:1275-9.
49. Lorico A, Rappa G, Finch RA, Yang D, Flavell RA, Sartorelli AC. Disruption of the murine MRP (multidrug resistance protein) gene leads to increased sensitivity to etoposide (VP-16) and increased levels of glutathione. Cancer Res 1997;57:5238-42.
50. Renes J, de Vries EG, Nienhuis EF, Jansen PL, Müller M. ATP- and glutathione-dependent transport of chemotherapeutic drugs by the multidrug resistance protein MRP1. Br J Pharmacol 1999;126:681-8.
51. Jedlitschky G, Leier I, Buchholz U, Barnouin K, Kurz G, Keppler D. Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res 1996;56:988.
52. Bakos é, Evers R, Szakács G, Tusnády GE, Welker E, Szabó K, de Haas M, van Deemter L, Borst P, Váradi A, Sarkadi B. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J Biol Chem 1998;273:32167-75.
53. Westlake CJ, Cole SP, Deeley RG. Role of the NH(2)-terminal membrane spanning domain of multidrug resistance protein 1/ABCC1 in protein processing and trafficking. Mol Biol Cell 2005;16:2483-92.
54. Yang Y, Chen Q, Zhang JT. Structural and functional consequences of mutating cysteine residues in the amino terminus of human multidrug resistance-associated protein 1. J Biol Chem 2002;277:44268-77.
55. Chen Q, Yang Y, Li L, Zhang JT. The amino terminus of the human multidrug resistance transporter ABCC1 has a U-shaped folding with a gating function. J Biol Chem 2006;281:31152-63.
56. Yang Y, Mo W, Zhang JT. Role of transmembrane segment 5 and extracellular loop 3 in the homodimerization of human ABCC1. Biochemistry 2010;49:10854-61.
57. Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer 2012;31:58-72.
58. Ween MP, Armstrong MA, Oehler MK, Ricciardelli C. The role of ABC transporters in ovarian cancer progression and chemoresistance. Crit Rev Oncol Hematol 2015;96:220-56.
59. Hipfner DR, Deeley RG, Cole SP. Structural, mechanistic and clinical aspects of MRP1. Biochim Biophys Acta 1999;1461:359-76.
60. Haber M, Smith J, Bordow SB, Flemming C, Cohn SL, London WB, Marshall GM, Norris MD. Association of high-level MRP1 expression with poor clinical outcome in a large prospective study of primary neuroblastoma. J Clin Oncol 2006;24:1546-53.
61. Li XQ, Li J, Shi SB, Chen P, Yu LC, Bao QL. Expression of MRP1, BCRP, LRP and ERCC1 as prognostic factors in non-small cell lung cancer patients receiving postoperative cisplatin-based chemotherapy. Int J Biol Markers 2009;24:230-7.
62. Filipits M, Pohl G, Rudas M, Dietze O, Lax S, Grill R, Pirker R, Zielinski CC, Hausmaninger H, Kubista E, Samonigg H, Jakesz R. Clinical role of multidrug resistance protein 1 expression in chemotherapy resistance in early-stage breast cancer: the Austrian Breast and Colorectal Cancer Study Group. J Clin Oncol 2005;23:1161-8.
63. Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport - an update. AAPS J 2015;17:65-82.
64. Taipalensuu J, Törnblom H, Lindberg G, Einarsson C, Sjöqvist F, Melhus H, Garberg P, Sjöström B, Lundgren B, Artursson P. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther 2001;299:164-70.
65. Merino G, álvarez AI, Pulido MM, Molina AJ, Schinkel AH, Prieto JG. Breast cancer resistance protein (BCRP/ABCG2) transports fluoroquinolone antibiotics and affects their oral availability, pharmacokinetics, and milk secretion. Drug Metab Dispos 2006;34:690.
66. Seamon JA, Rugg CA, Emanuel S, Calcagno AM, Ambudkar SV, Middleton SA, Butler J, Borowski V, Greenberger LM. Role of the ABCG2 drug transporter in the resistance and oral bioavailability of a potent cyclin-dependent kinase/Aurora kinase inhibitor. Mol Cancer Ther 2006;5:2459.
67. Yamagata T, Kusuhara H, Morishita M, Takayama K, Benameur H, Sugiyama Y. Improvement of the oral drug absorption of topotecan through the inhibition of intestinal xenobiotic efflux transporter, breast cancer resistance protein, by excipients. Drug Metab Dispos 2007;35:1142.
68. Tanaka Y, Kitamura Y, Maeda K, Sugiyama Y. Quantitative analysis of the ABCG2 c.421C>A polymorphism effect on in vivo transport activity of breast cancer resistance protein (BCRP) using an intestinal absorption model. J Pharm Sci 2015;104:3039-48.
69. Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol 2002;20:2943-50.
70. Staud F, Pavek P. Breast cancer resistance protein (BCRP/ABCG2). Int J Biochem Cell Biol 2005;37:720-5.
71. Clark R, Kerr ID, Callaghan R. Multiple drugbinding sites on the R482G isoform of the ABCG2 transporter. Br J Pharmacol 2006;149:506-15.
72. Giri N, Agarwal S, Shaik N, Pan G, Chen Y, Elmquist WF. Substrate-dependent breast cancer resistance protein (BCRP1/ABCG2)-mediated interactions: consideration of multiple binding sites in in vitro assay design. Drug Metab Dispos 2009;37:560-70.
73. Muenster U, Grieshop B, Ickenroth K, Gnoth MJ. Characterization of substrates and inhibitors for the in vitro assessment of Bcrp mediated drug-drug interactions. Pharm Res 2008;25:2320-6.
74. Hazai E, Bikádi Z. Homology modeling of breast cancer resistance protein (ABCG2). J Struct Biol 2008;162:63-74.
75. Kage K, Tsukahara S, Sugiyama T, Asada S, Ishikawa E, Tsuruo T, Sugimoto Y. Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int J Cancer 2002;97:626-30.
76. Mo W, Zhang JT. Oligomerization of human ATP-binding cassette transporters and its potential significance in human disease. Expert Opin Drug Metab Toxicol 2009;5:1049-63.
77. Ni Z, Bikadi Z, Rosenberg MF, Mao Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2). Curr Drug Metab 2010;11:603-17.
78. Diestra JE, Scheffer GL, Català I, Maliepaard M, Schellens JHM, Scheper RJ, Germà-Lluch JR, Izquierdo MA. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J Pathol 2002;198:213-9.
79. Damiani D, Tiribelli M, Calistri E, Geromin A, Chiarvesio A, Michelutti A, Cavallin M, Fanin R. The prognostic value of P-glycoprotein (ABCB) and breast cancer resistance protein (ABCG2) in adults with de novo acute myeloid leukemia with normal karyotype. Haematologica 2006;91:825-8.
80. Plasschaert SL, van der Kolk DM, de Bont ES, Kamps WA, Morisaki K, Bates SE, Scheffer GL, Scheper RJ, Vellenga E, de Vries EG. The role of breast cancer resistance protein in acute lymphoblastic leukemia. Clin Cancer Res 2003;9:5171-7.
81. Stam RW, van den Heuvel-Eibrink MM, den Boer ML, Ebus ME, Janka-Schaub GE, Allen JD, Pieters R. Multidrug resistance genes in infant acute lymphoblastic leukemia: Ara-C is not a substrate for the breast cancer resistance protein. Leukemia 2003;18:78-83.
82. Robey RW, To KKK, Polgar O, Dohse M, Fetsch P, Dean M, Bates SE. ABCG2: a perspective. Adv Drug Deliv Rev 2009;61:3-13.
83. Li-Blatter X, Beck A, Seelig A. P-glycoprotein-ATPase modulation: the molecular mechanisms. Biophys J 2012;102:1383-93.
84. Maki N, Moitra K, Ghosh P, Dey S. Allosteric modulation bypasses the requirement for ATP hydrolysis in regenerating low affinity transition state conformation of human P-glycoprotein. J Biol Chem 2006;281:10769-77.
85. Romsicki Y, Sharom FJ. The membrane lipid environment modulates drug interactions with the P-glycoprotein multidrug transporter. Biochemistry 1999;38:6887-96.
86. Aliabadi HM, Landry B, Mahdipoor P, Hsu CY, Uludağ H. Effective down-regulation of breast cancer resistance protein (BCRP) by siRNA delivery using lipid-substituted aliphatic polymers. Eur J Pharm Biopharm 2012;81:33-42.
87. Gomes MJ, Kennedy PJ, Martins S, Sarmento B. Delivery of siRNA silencing P-gp in peptide-functionalized nanoparticles causes efflux modulation at the blood-brain barrier. Nanomedicine (Lond) 2017; doi: 10.2217/nnm-2017-0023. doi:10.2217/nnm-2017-0023.
88. Fisher M, Abramov M, Van Aerschot A, Xu D, Juliano RL, Herdewijn P. Inhibition of MDR1 expression with altritol-modified siRNAs. Nucleic Acids Res 2007;35:1064-74.
89. Lam JK, Chow MY, Zhang Y, Leung SW. siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucleic Acids 2015;4:e252.
90. Cuiffo BG, Campagne A, Bell GW, Lembo A, Orso F, Lien EC, Bhasin MK, Raimo M, Hanson SE, Marusyk A, El-Ashry D, Hematti P, Polyak K, Mechta-Grigoriou F, Mariani O, Volinia S, Vincent-Salomon A, Taverna D, Karnoub AE. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell 2014;15:762-74.
91. Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: multi-targeted or single-targeted? World J Clin Oncol 2011;2:80-93.
92. Liu Y, Zhu W, Zhang J, Huang J, Zhao Y, Zhang W, Han B, Yao Y, Jiang L, Li S. The evaluation of efficacy and safety of sunitinib on EGFR-TKI pretreated advanced non-small cell lung cancer patients in China. Clin Respir J 2014;8:206-12.
93. Na F, Zhang J, Deng L, Zhou X, Zhou L, Zou B, Yu M, Li Y, Xue J, Liu Y. Continuation of tyrosine kinase inhibitor is associated with survival benefit in NSCLC patients with exon 19 deletion after solitary progression. J Cancer 2017;8:3682-8.
94. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28-39.
95. Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 2015;36:422-39.
96. Rask-Andersen M, Zhang J, Fabbro D, Schiöth HB. Advances in kinase targeting: current clinical use and clinical trials. Trends Pharmacol Sci 2014;35:604-20.
97. Kathawala RJ, Gupta P, Ashby CR, Chen ZS. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat 2015;18:1-17.
98. List AF, Kopecky KJ, Willman CL, Head DR, Persons DL, Slovak ML, Dorr R, Karanes C, Hynes HE, Doroshow JH, Shurafa M, Appelbaum FR. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood 2001;98:3212-20.
99. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006;5:219-34.
100. Callaghan R, Luk F, Bebawy M. Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy? Drug Metab Dispos 2014;42:623-31.
101. List AF, Kopecky KJ, Willman CL, Head DR, Slovak ML, Douer D, Dakhil SR, Appelbaum FR. Cyclosporine inhibition of P-glycoprotein in chronic myeloid leukemia blast phase. Blood 2002;100:1910-2.
102. Abraham J, Edgerly M, Wilson R, Chen C, Rutt A, Bakke S, Robey R, Dwyer A, Goldspiel B, Balis F, Van Tellingen O, Bates SE, Fojo T. A phase I study of the P-glycoprotein antagonist tariquidar in combination with vinorelbine. Clin Cancer Res 2009;15:3574-82.
103. Gerrard G, Payne E, Baker R, Jones D, Potter M, Prentice H, Ethell M, McCullough H, Burgess M, Mehta A, Ganeshaguru K. Clinical effects and P-glycoprotein inhibition in patients with acute myeloid leukemia treated with zosuquidar trihydrochloride, daunorubicin and cytarabine. Haematologica 2004;89:782-90.
104. Sandler A, Gordon M, de Alwis DP, Pouliquen I, Green L, Marder P, Chaudhary A, Fife K, Battiato L, Sweeney C, Jordan C, Burgess M, Slapak CA. A phase I trial of a potent P-glycoprotein inhibitor, zosuquidar trihydrochloride (LY335979), administered intravenously in combination with doxorubicin in patients with advanced malignancy. Clin Cancer Res 2004;10:3265-72.
105. Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM, Rowe JM, Lazarus HM, Luger S, Tallman MS. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood 2010;116:4077-85.
106. Kühnle M, Egger M, Müller C, Mahringer A, Bernhardt G, Fricker G, König B, Buschauer A. Potent and selective inhibitors of breast cancer resistance protein (ABCG2) derived from the P-glycoprotein (ABCB1) modulator tariquidar. J Med Chem 2009;52:1190-7.
107. Li XQ, Wang L, Lei Y, Hu T, Zhang FL, Cho CH, To KK. Reversal of P-gp and BCRP-mediated MDR by tariquidar derivatives. Eur J Med Chem 2015;101:560-72.
108. Kakarla P, Inupakutika M, Devireddy AR, Gunda SK, Willmon TM, Ranjana K, Shrestha U, Ranaweera I, Hernandez AJ, Barr S, Varela MF. 3D-QSAR and contour map analysis of tariquidar analogues as multidrug resistance protein-1 (MRP1) inhibitors. Int J Pharm Sci Res 2016;7:554-72.
109. Kannan P, Telu S, Shukla S, Ambudkar SV, Pike VW, Halldin C, Gottesman MM, Innis RB, Hall MD. The "specific" P-glycoprotein inhibitor tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem Neurosci 2011;2:82-9.
110. Martin C, Berridge G, Mistry P, Higgins C, Charlton P, Callaghan R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br J Pharmacol 1999;128:403-11.
111. Bankstahl JP, Bankstahl M, Römermann K, Wanek T, Stanek J, Windhorst AD, Fedrowitz M, Erker T, Müller M, Löscher W, Langer O, Kuntner C. Tariquidar and elacridar are dose-dependently transported by P-glycoprotein and BCRP at the blood-brain barrier: a small-animal positron emission tomography and in vitro study. Drug Metab Dispos 2013;41:754-62.
112. Weidner LD, Fung KL, Kannan P, Moen JK, Kumar JS, Mulder J, Innis RB, Gottesman MM, Hall MD. Tariquidar is an inhibitor and not a substrate of human and mouse P-glycoprotein. Drug Metab Dispos 2016;44:275-82.
113. Mross K, Bohn C, Edler L, Jonat W, Queisser W, Heidemann E, Goebel M, Hossfeld DK. Randomized phase II study of single-agent epirubicin +/- verapamil in patients with advanced metastatic breast cancer. An AIO clinical trial. Arbeitsgemeinschaft Internistische Onkologie of the German Cancer Society. Ann Oncol 1993;4:45-50.
114. Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist 2003;8:411-24.
115. Porcelli L, Lemos C, Peters GJ, Paradiso A, Azzariti A. Intracellular trafficking of MDR transporters and relevance of SNPs. Curr Top Med Chem 2009;9:197-208.
116. Follit CA, Brewer FK, Wise JG, Vogel PD. In silico identified targeted inhibitors of P-glycoprotein overcome multidrug resistance in human cancer cells in culture. Pharmacol Res Perspect 2015;3:e00170.
117. Brewer FK, Follit CA, Vogel PD, Wise JG. In silico screening for inhibitors of p-glycoprotein that target the nucleotide binding domains. Mol Pharmacol 2014;86:716-26.
118. Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci Rep 2018;8:967.
119. Zinzi L, Capparelli E, Cantore M, Contino M, Leopoldo M, Colabufo NA. Small and innovative molecules as new strategy to revert MDR. Front Oncol 2014;4:2.
120. Yang X, Iyer AK, Singh A, Milane L, Choy E, Hornicek FJ, Amiji MM, Duan Z. Cluster of differentiation 44 targeted hyaluronic acid based nanoparticles for MDR1 siRNA delivery to overcome drug resistance in ovarian cancer. Pharm Res 2015;32:2097-109.
121. Ganesh S, Iyer AK, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials 2013;34:3489-502.
122. Wu D, Li XS, Meng XN, Yan J, Zong ZH. MicroRNA-873 mediates multidrug resistance in ovarian cancer cells by targeting ABCB1. Tumour Biol 2016;37:10499-506.
123. Zhu H, Wu H, Liu X, Evans BR, Medina DJ, Liu CG, Yang JM. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem Pharmacol 2008;76:582-8.
124. Da Silva CG, Peters GJ, Ossendorp F, Cruz LJ. The potential of multi-compound nanoparticles to bypass drug resistance in cancer. Cancer Chemother Pharmacol 2017;80:881-94.
125. Iwamoto T. Clinical application of drug delivery systems in cancer chemotherapy: review of the efficacy and side effects of approved drugs. Biol Pharm Bull 2013;36:715-8.
126. Yamamoto Y, Kawano I, Iwase H. Nab-paclitaxel for the treatment of breast cancer: efficacy, safety, and approval. OncoTargets Ther 2011;4:123-36.
127. Jin X, Zhou B, Xue L, San W. Soluplus® micelles as a potential drug delivery system for reversal of resistant tumor. Biomed Pharmacother 2015;69:388-95.
129. De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine 2008;3:133-49.
130. Zhou Y, Hong L. Prediction value of miR-483 and miR-214 in prognosis and multidrug resistance of esophageal squamous cell carcinoma. Genet Test Mol Biomark 2013;17:470-4.
131. Li Z, Hu S, Wang J, Cai J, Xiao L, Yu L, Wang Z. MiR-27a modulates MDR1/P-glycoprotein expression by targeting HIPK2 in human ovarian cancer cells. Gynecol Oncol 2010;119:125-30.
132. Chen Z, Ma T, Huang C, Zhang L, Lv X, Xu T, Hu T, Li J. MiR-27a modulates the MDR1/P-glycoprotein expression by inhibiting FZD7/β-catenin pathway in hepatocellular carcinoma cells. Cell Signal 2013;25:2693-701.
133. Feng DD, Zhang H, Zhang P, Zheng YS, Zhang XJ, Han BW, Luo XQ, Xu L, Zhou H, Qu LH, Chen YQ. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J Cell Mol Med 2011;15:2164-75.
134. Hong L, Han Y, Zhang H, Li M, Gong T, Sun L, Wu K, Zhao Q, Fan D. The prognostic and chemotherapeutic value of miR-296 in esophageal squamous cell carcinoma. Ann Surg 2010;251:1056-63.
135. Lee SD, Osei-Twum JA, Wasan KM. Dose-dependent targeted suppression of P-glycoprotein expression and function in Caco-2 cells. Mol Pharm 2013;10:2323-30.
136. Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol 2012;180:2490-503.
137. Lin CJ, Gong HY, Tseng HC, Wang WL, Wu JL. miR-122 targets an anti-apoptotic gene, Bcl-w, in human hepatocellular carcinoma cell lines. Biochem Biophys Res Commun 2008;375:315-20.
138. Xu Y, Ohms SJ, Li Z, Wang Q, Gong G, Hu Y, Mao Z, Shannon MF, Fan JY. Changes in the expression of miR-381 and miR-495 are inversely associated with the expression of the MDR1 gene and development of multi-drug resistance. PLoS One 2013;8:e82062.
139. Yang T, Zheng ZM, Li XN, Li ZF, Wang Y, Geng YF, Bai L, Zhang XB. MiR-223 modulates multidrug resistance via downregulation of ABCB1 in hepatocellular carcinoma cells. Exp Biol Med (Maywood) 2013;238:1024-32.
140. Munoz JL, Bliss SA, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Delivery of functional anti-miR-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Mol Ther Nucleic Acids 2013;2:e126.
141. Nobili S, Landini I, Mazzei T, Mini E. Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med Res Rev 2012;32:1220-62.
142. Stupp R, Tosoni A, Bromberg JEC, Hau P, Campone M, Gijtenbeek J, Frenay M, Breimer L, Wiesinger H, Allgeier A, van den Bent MJ, Bogdahn U, van der Graaf W, Yun HJ, Gorlia T, Lacombe D, Brandes AA. Sagopilone (ZK-EPO, ZK 219477) for recurrent glioblastoma. A phase II multicenter trial by the European Organisation for Research and Treatment of Cancer (EORTC) Brain Tumor Group. Ann Oncol 2011;22:2144-9.
143. Lee FY, Borzilleri R, Fairchild CR, Kim SH, Long BH, Reventos-Suarez C, Vite GD, Rose WC, Kramer RA. BMS-247550: a novel epothilone analog with a mode of action similar to paclitaxel but possessing superior antitumor efficacy. Clin Cancer Res 2001;7:1429-37.
144. Lechleider RJ, Kaminskas E, Jiang X, Aziz R, Bullock J, Kasliwal R, Harapanhalli R, Pope S, Sridhara R, Leighton J, Booth B, Dagher R, Justice R, Pazdur R. Ixabepilone in combination with capecitabine and as monotherapy for treatment of advanced breast cancer refractory to previous chemotherapies. Clin Cancer Res 2008;14:4378-84.
145. Puhalla S, Brufsky A. Ixabepilone: a new chemotherapeutic option for refractory metastatic breast cancer. Biol Targets Ther 2008;2:505-15.
146. Sparano JA, Vrdoljak E, Rixe O, Xu B, Manikhas A, Medina C, Ventilari Da Costa SC, Ro J, Rubio G, Rondinon M, Perez Manga G, Peck R, Poulart V, Conte P. Randomized phase III trial of ixabepilone plus capecitabine versus capecitabine in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol 2010;28:3256-63.
147. Mitchell D, Bergendahl G, Ferguson W, Roberts W, Higgins T, Ashikaga T, DeSarno M, Kaplan J, Kraveka J, Eslin D, Vander Werff A, Hanna GK, Sholler GL. A phase 1 trial of TPI 287 as a single agent and in combination with temozolomide in patients with refractory or recurrent neuroblastoma or medulloblastoma. Pediatr Blood Cancer 2016;69:39-46.
148. Dumontet C, Jordan MA, Lee FF. Ixabepilone: targeting betaIII-tubulin expression in taxane-resistant malignancies. Mol Cancer Ther 2009;8:17-25.
149. Shen H, Lee FY, Gan J. Ixabepilone, a novel microtubule-targeting agent for breast cancer, is a substrate for P-glycoprotein (P-gp/MDR1/ABCB1) but not breast cancer resistance protein (BCRP/ABCG2). J Pharmacol Exp Ther 2011;337:423-32.
150. Yang CP, Verdier-Pinard P, Wang F, Lippaine-Horvath E, He L, Li D, Höfle G, Ojima I, Orr GA, Horwitz SB. A highly epothilone B-resistant A549 cell line with mutations in tubulin that confer drug dependence. Mol Cancer Ther 2005;4:987-95.
151. Wangari-Talbot J, Hopper-Borge E. Drug resistance mechanisms in non-small cell lung carcinoma. J Can Res Updat 2014;2:265-82.
152. Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol Sci 2009;30:546-56.
153. Bergman AM, Munch-Petersen B, Jensen PB, Sehested M, Veerman G, Voorn DA, Smid K, Pinedo HM, Peters GJ. Collateral sensitivity to gemcitabine (2',2'-difluorodeoxycytidine) and cytosine arabinoside of daunorubicin- and VM-26-resistant variants of human small cell lung cancer cell lines. Biochem Pharmacol 2001;61:1401-8.
154. Nakagawa-Goto K, Bastow KF, Ohkoshi E, Morris-Natschke SL, Lee KH. Antitumor agents 291 expanded B-Ring modification study of 6,8,8-triethyl desmosdumotin B analogues as multidrug-resistance selective agents. Med Chem 2011;1:1000101.
155. Hall MD, Brimacombe KR, Varonka MS, Pluchino KM, Monda JK, Li J, Walsh MJ, Boxer MB, Warren TH, Fales HM, Gottesman MM. Synthesis and structure-activity evaluation of isatin-β-thiosemicarbazones with improved selective activity towards multidrug-resistant cells expressing P-glycoprotein. J Med Chem 2011;54:5878-89.
156. Vine KL, Belfiore L, Jones L, Locke JM, Wade S, Minaei E, Ranson M. N-alkylated isatins evade P-gp mediated efflux and retain potency in MDR cancer cell lines. Heliyon 2016;2:e00060.
157. Bergman AM, Pinedo HM, Talianidis I, Veerman G, Loves WJ, van der Wilt CL, Peters GJ. Increased sensitivity to gemcitabine of P-glycoprotein and multidrug resistance-associated protein-overexpressing human cancer cell lines. Br J Cancer 2003;88:1963-70.
158. Bergman AM, Pinedo HM, Peters GJ. Steroids affect collateral sensitivity to gemcitabine of multidrug-resistant human lung cancer cells. Eur J Pharmacol 2001;416:19-24.
159. Borner MM, Schoffski P, de Wit R, Caponigro F, Comella G, Sulkes A, Greim G, Peters GJ, van der Born K, Wanders J, de Boer RF, Martin C, Fumoleau P. Patient preference and pharmacokinetics of oral modulated UFT versus intravenous fluorouracil and leucovorin: a randomised crossover trial in advanced colorectal cancer. Eur J Cancer 2002;38:349-58.
160. Liu G, Franssen E, Fitch MI, Warner E. Patient preferences for oral versus intravenous palliative chemotherapy. J Clin Oncol 1997;15:110-5.
161. Grant DS, Williams TL, Zahaczewsky M, Dicker AP. Comparison of antiangiogenic activities using paclitaxel (taxol) and docetaxel (taxotere). Int J Cancer 2003;104:121-9.
162. Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 2004;4:423-36.
163. Sparreboom A, van Asperen J, Mayer U, Schinkel AH, Smit JW, Meijer DK, Borst P, Nooijen WJ, Beijnen JH, van Tellingen O. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci U S A 1997;94:2031-5.
164. van Asperen J, van Tellingen O, van der Valk MA, Rozenhart M, Beijnen JH. Enhanced oral absorption and decreased elimination of paclitaxel in mice cotreated with cyclosporin A. Clin Cancer Res 1998;4:2293-7.
165. Bardelmeijer HA, Ouwehand M, Beijnen JH, Schellens JH, van Tellingen O. Efficacy of novel P-glycoprotein inhibitors to increase the oral uptake of paclitaxel in mice. Invest New Drugs 2004;22:219-29.
166. Bardelmeijer HA, Beijnen JH, Brouwer KR, Rosing H, Nooijen WJ, Schellens JHM, van Tellingen O. Increased oral bioavailability of paclitaxel by GF120918 in mice through selective modulation of P-glycoprotein. Clin Cancer Res 2000;6:4416-21.
167. Koolen SL, Beijnen JH, Schellens JH. Intravenous-to-oral switch in anticancer chemotherapy: a focus on docetaxel and paclitaxel. Clin Pharmacol Ther 2010;87:126-9.
168. Shukla S, Ohnuma S, Ambudkar SV. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr Drug Targets 2011;12:621-30.
169. Wils P, Phung-Ba V, Warnery A, Lechardeur D, Raeissi S, Hidalgo IJ, Scherman D. Polarized transport of docetaxel and vinblastine mediated by P-glycoprotein in human intestinal epithelial cell monolayers. Biochem Pharmacol 1994;48:1528-30.
170. Jibodh RA, Lagas JS, Nuijen B, Beijnen JH, Schellens JH. Taxanes: old drugs, new oral formulations. New Front Breast Cancer Treat 2013;717:40-6.
171. Rezonja R, Knez L, Cufer T, Mrhar A. Oral treatment with etoposide in small cell lung cancer - dilemmas and solutions. Radiol Oncol 2013;47:1-13.
172. Hamada H, Tsuruo T. Functional role for the 170- to 180-kDa glycoprotein specific to drug-resistant tumor cells as revealed by monoclonal antibodies. Proc Natl Acad Sci U S A 1986;83:7785-9.
173. Pearson JW, Fogler WE, Volker K, Usui N, Goldenberg SK, Gruys E, Riggs CW, Komschlies K, Wiltrout RH, Tsuruo T. Reversal of drug resistance in a human colon cancer xenograft expressing MDR1 complementary DNA by in vivo administration of MRK-16 monoclonal antibody. J Natl Cancer Inst 1991;83:1386-91.
174. Naito M, Tsuge H, Kuroko C, Koyama T, Tomida A, Tatsuta T, Heike Y, Tsuruo T. Enhancement of cellular accumulation of cyclosporine by anti-P-glycoprotein monoclonal antibody MRK-16 and synergistic modulation of multidrug resistance. J Natl Cancer Inst 1993;85:311-6.
175. Naito M, Tsuruo T. [18]Monoclonal antibodies to P-glycoprotein: preparation and applications to basic and clinical research. Methods Enzymol 1998;292:258-65.
176. Cole SP. Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol 2014;54:95-117.
177. Wang S, Tan X, Li S, Zhou Y, Geng P, Hua A, Deng A, Yu Z. Indomethacin-based stimuli-responsive micelles combined with paclitaxel to overcome multidrug resistance. Oncotarget 2017;8:111281-94.
178. Wan Y, Apostolou S, Dronov R, Kuss B, Voelcker NH. Cancer-targeting siRNA delivery from porous silicon nanoparticles. Nanomedicine (Lond) 2014;9:2309-21.
179. Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, Smith NR, James NH, Dukes M, Curwen JO, Chester R, Jackson JA, Boffey SJ, Kilburn LL, Barnett S, Richmond GHP, Wadsworth PF, Walker M, Bigley AL, Taylor ST, Cooper L, Beck S, Jürgensmeier JM, Ogilvie DJ. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res 2005;65:4389-400.
180. Tao LY, Liang YJ, Wang F, Chen LM, Yan YY, Dai CL, Fu LW. Cediranib (recentin, AZD2171) reverses ABCB1- and ABCC1-mediated multidrug resistance by inhibition of their transport function. Cancer Chemother Pharmacol 2009;64:961-9.
181. Zheng L, Wang F, Li Y, Zhang X, Chen L, Liang Y, Dai C, Yan Y, Tao L, Mi Y, Yang A, To KKW, Fu L. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS One 2009;4:e5172.
182. Mi Y, Lou L. ZD6474 reverses multidrug resistance by directly inhibiting the function of P-glycoprotein. Br J Cancer 2007;97:934-40.
183. Hegedus T, Orfi L, Seprodi A, Váradi A, Sarkadi B, Kéri G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta 2002;1587:318-25.
184. Leier I, Jedlitschky G, Buchholz U, Cole SP, Deeley RG, Keppler D. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem 1994;269:27807-10.
185. Minderman H, O'Loughlin KL, Pendyala L, Baer MR. VX-710 (Biricodar) increases drug retention and enhances chemosensitivity in resistant cells overexpressing P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Clin Cancer Res 2004;10:1826-34.
186. Katayama R, Koike S, Sato S, Sugimoto Y, Tsuruo T, Fujita N. Dofequidar fumarate sensitizes cancer stem-like side population cells to chemotherapeutic drugs by inhibiting ABCG2/BCRP-mediated drug export. Cancer Sci 2009;100:2060-8.
187. Robey RW, Shukla S, Finley EM, Oldham RK, Barnett D, Ambudkar SV, Fojo T, Bates SE. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1®. Biochem Pharmacol 2008;75:1302-12.
188. Ji L, Yang S, Li S, Liu S, Tang S, Liu Z, Meng X, Yu S. A novel triazolonaphthalimide induces apoptosis and inhibits tumor growth by targeting DNA and DNA-associated processes. Oncotarget 2017;8:37394-408.
189. Saeki T, Nomizu T, Toi M, Ito Y, Noguchi S, Kobayashi T, Asaga T, Minami H, Yamamoto N, Aogi K, Ikeda T, Ohashi Y, Sato W, Tsuruo T. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J Clin Oncol 2007;25:411-7.
190. Cole SP. Multidrug resistance protein 1 (MRP1, ABCC1), a "multitasking" ATP-binding cassette (ABC) transporter. J Biol Chem 2014;289:30880-8.
191. Trompier D, Chang XB, Barattin R, du Moulinet D'Hardemare A, Di Pietro A, Baubichon-Cortay H. Verapamil and its derivative trigger apoptosis through glutathione extrusion by multidrug resistance protein MRP1. Cancer Res 2004;64:4950-6.
192. de Groot DJA, van der Deen M, Le TK, Regeling A, de Jong S, de Vries EG. Indomethacin induces apoptosis via a MRP1-dependent mechanism in doxorubicin-resistant small-cell lung cancer cells overexpressing MRP1. Br J Cancer 2007;97:1077-83.
193. Ahmed-Belkacem A, Pozza A, Macalou S, Pérez-Victoria JM, Boumendjel A, Di Pietro A. Inhibitors of cancer cell multidrug resistance mediated by breast cancer resistance protein (BCRP/ABCG2). Anticancer Drugs 2006;17:239-43.
194. Rabindran SK, He H, Singh M, Brown E, Collins KI, Annable T, Greenberger LM. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res 1998;58:5850-8.
195. Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, Schellens JH, Koomen GJ, Schinkel AH. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther 2002;1:417-25.
196. Pick A, Klinkhammer W, Wiese M. Specific inhibitors of the breast cancer resistance protein (BCRP). Chem Med Chem 2010;5:1498-505.
197. Peng H, Dong Z, Qi J, Yang Y, Liu Y, Li Z, Xu J, Zhang JT. A novel two mode-acting inhibitor of ABCG2-mediated multidrug transport and resistance in cancer chemotherapy. PLoS One 2009;4:e5676.
198. Komeili-Movahhed T, Fouladdel S, Barzegar E, Atashpour S, Hossein Ghahremani M, Nasser Ostad S, Madjd Z, Azizi E. PI3K/Akt inhibition and down-regulation of BCRP re-sensitize MCF7 breast cancer cell line to mitoxantrone chemotherapy. Iran J Basic Med Sci 2015;18:472-7.
199. Singh MS, Juvale K, Wiese M, Lamprecht A. Evaluation of dual P-gp-BCRP inhibitors as nanoparticle formulation. Eur J Pharm Sci 2015;77:1-8.
200. Li WQ, Li YM, Tao BB, Lu YC, Hu GH, Liu HM, He J, Xu Y, Yu HY. Downregulation of ABCG2 expression in glioblastoma cancer stem cells with miRNA-328 may decrease their chemoresistance. Med Sci Monit 2010;16:HY27-30.
201. Li X, Pan YZ, Seigel GM, Hu ZH, Huang M, Yu AM. Breast cancer resistance protein BCRP/ABCG2 regulatory microRNAs (hsa-miR-328, -519c and -520h) and their differential expression in stem-like ABCG2+ cancer cells. Biochem Pharmacol 2011;81:783-92.
202. Turrini E, Haenisch S, Laechelt S, Diewock T, Bruhn O, Cascorbi I. MicroRNA profiling in K-562 cells under imatinib treatment: influence of miR-212 and miR-328 on ABCG2 expression. Pharmacogenet Genomics 2012;22:198-205.
203. Jiao X, Zhao L, Ma M, Bai X, He M, Yan Y, Wang Y, Chen Q, Zhao X, Zhou M, Cui Z, Zheng Z, Wang E, Wei M. MiR-181a enhances drug sensitivity in mitoxantone-resistant breast cancer cells by targeting breast cancer resistance protein (BCRP/ABCG2). Breast Cancer Res Treat 2013;139:717-30.
204. Ma MT, He M, Wang Y, Jiao XY, Zhao L, Bai XF, Yu ZJ, Wu HZ, Sun ML, Song ZG, Wei MJ. MiR-487a resensitizes mitoxantrone (MX)-resistant breast cancer cells (MCF-7/MX) to MX by targeting breast cancer resistance protein (BCRP/ABCG2). Cancer Lett 2013;339:107-15.
205. Wang Y, Zhao L, Xiao Q, Jiang L, He M, Bai X, Ma M, Jiao X, Wei M. miR-302a/b/c/d cooperatively inhibit BCRP expression to increase drug sensitivity in breast cancer cells. Gynecol Oncol 2016;141:592-601.
206. Zhang L, Guo X, Zhang D, Fan Y, Qin L, Dong S, Zhang L. Upregulated miR-132 in Lgr5+gastric cancer stem cell-like cells contributes to cisplatin-resistance via SIRT1/CREB/ABCG2 signaling pathway. Mol Carcinog 2017;56:2022-34.
207. Chen B, Zhang D, Kuai J, Cheng M, Fang X, Li G. Upregulation of miR-199a/b contributes to cisplatin resistance via Wnt/β-catenin-ABCG2 signaling pathway in ALDHA1+colorectal cancer stem cells. Tumour Biol 2017;39:1010428317715155.
208. Shukla S, Chen ZS, Ambudkar SV. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist Updat 2012;15:70-80.
209. Azzariti A, Porcelli L, Simone GM, Quatrale AE, Colabufo NA, Berardi F, Perrone R, Zucchetti M, D'Incalci M, Xu JM, Paradiso A. Tyrosine kinase inhibitors and multidrug resistance proteins: interactions and biological consequences. Cancer Chemother Pharmacol 2010;65:335-46.
210. Ozvegy-Laczka C, Hegedus T, Várady G, Ujhelly O, Schuetz JD, Váradi A, Kéri G, Orfi L, Német K, Sarkadi B. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol 2004;65:1485-95.
211. Veronese ML, Sun W, Giantonio B, Berlin J, Shults J, Davis L, Haller DG, O'Dwyer PJ. A phase II trial of gefitinib with 5-fluorouracil, leucovorin, and irinotecan in patients with colorectal cancer. Br J Cancer 2005;92:1846-9.
212. Deenik W, Janssen JJ, van der Holt B, Verhoef GE, Smit WM, Kersten MJ, Daenen SM, Verdonck LF, Ferrant A, Schattenberg AV, Sonneveld P, van Marwijk Kooy M, Wittebol S, Willemze R, Wijermans PW, Beverloo HB, Löwenberg B, Valk PJ, Ossenkoppele GJ, Cornelissen JJ. Efficacy of escalated imatinib combined with cytarabine in newly diagnosed patients with chronic myeloid leukemia. Haematologica 2010;95:914-21.
213. Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol 2003;21:2237-46.
214. Giaccone G, González-Larriba JL, van Oosterom AT, Alfonso R, Smit EF, Martens M, Peters GJ, van der Vijgh WJF, Smith R, Averbuch S, Fandi A. Combination therapy with gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, gemcitabine and cisplatin in patients with advanced solid tumors. Ann Oncol 2004;15:831-8.
215. Stewart CF, Leggas M, Schuetz JD, Panetta JC, Cheshire PJ, Peterson J, Daw N, Jenkins JJ, Gilbertson R, Germain GS, Harwood FC, Houghton PJ. Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Res 2004;64:7491-9.
216. Leggas M, Panetta JC, Zhuang Y, Schuetz JD, Johnston B, Bai F, Sorrentino B, Zhou S, Houghton PJ, Stewart CF. Gefitinib modulates the function of multiple ATP-binding cassette transporters in vivo. Cancer Res 2006;66:4802-7.
218. Leslie EM, Mao Q, Oleschuk CJ, Deeley RG, Cole SP. Modulation of multidrug resistance protein 1 (MRP1/ABCC1) transport and atpase activities by interaction with dietary flavonoids. Mol Pharmacol 2001;59:1171-80.
219. Alvarez AI, Real R, Pérez M, Mendoza G, Prieto JG, Merino G. Modulation of the activity of ABC transporters (P-glycoprotein, MRP2, BCRP) by flavonoids and drug response. J Pharm Sci 2010;99:598-617.
220. Cooray HC, Janvilisri T, van Veen HW, Hladky SB, Barrand MA. Interaction of the breast cancer resistance protein with plant polyphenols. Biochem Biophys Res Commun 2004;317:269-75.
221. Pan L, Hu H, Wang X, Yu L, Jiang H, Chen J, Lou Y, Zeng S. Inhibitory effects of neochamaejasmin B on P-glycoprotein in MDCK-hMDR1 cells and molecular docking of NCB binding in P-glycoprotein. Molecules 2015;20:2931-48.
222. Forester SC, Waterhouse AL. Metabolites are key to understanding health effects of wine polyphenolics. J Nutr 2009;139:S1824-31.
223. Bohn T, McDougall GJ, Alegría A, Alminger M, Arrigoni E, Aura AM, Brito C, Cilla A, El SN, Karakaya S, Martínez-Cuesta MC, Santos CN. Mind the gap-deficits in our knowledge of aspects impacting the bioavailability of phytochemicals and their metabolites--a position paper focusing on carotenoids and polyphenols. Mol Nutr Food Res 2015;59:1307-23.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Jaramillo AC, Saig FA, Cloos J, Jansen G, Peters GJ. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance?. Cancer Drug Resist 2018;1:6-29. http://dx.doi.org/10.20517/cdr.2018.02
AMA Style
Jaramillo AC, Saig FA, Cloos J, Jansen G, Peters GJ. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance?. Cancer Drug Resistance. 2018; 1(1): 6-29. http://dx.doi.org/10.20517/cdr.2018.02
Chicago/Turabian Style
Jaramillo, Adrian C., Farah Al Saig, Jacqueline Cloos, Gerrit Jansen, Godefridus J. Peters. 2018. "How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance?" Cancer Drug Resistance. 1, no.1: 6-29. http://dx.doi.org/10.20517/cdr.2018.02
ACS Style
Jaramillo, AC.; Saig FA.; Cloos J.; Jansen G.; Peters GJ. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance?. Cancer Drug Resist. 2018, 1, 6-29. http://dx.doi.org/10.20517/cdr.2018.02
About This Article
Copyright

Data & Comments
Data


 Cite This Article 53 clicks
Cite This Article 53 clicks

 Like This Article 9
likes
Like This Article 9
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.