
Reduction of mitomycin C resistance in human bladder cancer T24 cells by knocking-down ras oncogene

Abstract
Aim: Mitomycin C (MMC) is a commonly used as intravesical treatment for superficial bladder cancer. However, its role in combination with ras inhibition has not been investigated. The aim of this study was to explore the role of ras in MMC-induced apoptosis in T24 bladder cancer cells and to determine the efficacy of combination therapy in vitro.
Methods: We measured the effects of various doses of MMC on apoptosis induction as well as on ras, ERK and Ki-67 protein expression by T24 cell line using immunocytochemistry, flow cytometry and Western blotting. We also tested the effect of siRNA on ras employed singly or in combination with MMC.
Results: T24 cells expressed high level of ras protein. MMC treatment increased the level of ras and ERK protein expression after 24 h, and decreased these levels after 72 h. Ras siRNA (100 nmol/L) caused massive apoptosis associated with a marked decrease in ras expression in T24 cells. When combined with low doses of MMC, ras siRNA (50 nmol/L) sensitized T24 cells to apoptosis and decreased their expression of ras. The effect of combined therapy was higher than that of either compound used alone. Expression levels of ERK, a downstream target of ras, declined following combination therapy.
Conclusion: Ras siRNA in combination with low dose MMC is a possible treatment strategy for patients with ras-positive bladder tumors.
Keywords
Introduction
Bladder cancer is one of the leading causes of cancer-related death in the world[1]. Despite the increasing number of studies in recent years, the recurrence and progression of bladder cancer remains a challenge. Therefore, new strategies are necessary. The standard treatment for non-muscle-invasive transitional cell carcinoma of the bladder (TCCB) is transurethral resection and adjuvant intravesical chemotherapy or immunotherapy[2,3]. However, clinical effectiveness of mitomycin C (MMC) is often limited by the emergence of drug-resistant tumor cells[4,5]. Approximately 43% of the non-muscle-invasive bladder cancer relapses and about 8% progress to muscle-invasive disease[6].
A substantial body of literature has documented the association of bladder cancer and tumor resistance to chemotherapy with dysregulated production of the signal transducer protein ras[7]. This association was reported in the T24 bladder carcinoma cell line, where the first human oncogene was detected[8]. Subsequent studies have underscored the crucial role of mutated ras proteins in bladder cancer. Mutations in RAS genes can lead to production of a ras protein without intrinsic GTPase activity. Dysregulation of a variety of other upstream and downstream components of cell cycle progression, proliferation and apoptosis pathways ultimately leads to uninterrupted cell growth and carcinogenesis[9]. Overexpression of at least one of the three ras canonical proteins was shown in 77% of the analyzed tumors, with K-ras and N-ras primarily overexpressed in bladder carcinoma. H-ras was more frequently overexpressed in transitional cell carcinomas[10]. Even a single nucleotide polymorphism in the H-ras locus was associated with a higher risk of developing bladder carcinomas and with advanced, more aggressive types of cancer[11].
Mitogen-activated protein kinases (MAPKs) are an evolutionary conserved family of protein kinases[12,13]. In multicellular organisms, there are three well-characterized subfamilies of MAPKs. These MAPKs include the extracellular signal-regulated kinases, ERK1 and ERK2, the c-Jun NH2-terminal kinases, and the four p38 enzymes. The ERK1 and ERK2 pathway is one of the best characterized and the most strongly related to human cancer among MAPK pathways[12]. ERK-1 and ERK-2 are ubiquitously expressed and are involved in cell proliferation, differentiation, apoptosis, and angiogenesis. Oncogenic ras persistently activates the ERK-1/ERK-2 pathway, leading to increased proliferation rate and resistance of tumor cells[13].
Ras inhibition was employed as a treatment strategy to control cell proliferation, to induce apoptosis, and to increase the sensitivity of tumor cells to various tumor cell-killing agents[14]. The inhibition was targeted at many levels including RAS gene, ras mRNA (by small interfering RNA or siRNA) or by blocking ras protein anchorage to cell membranes. In addition, upstream activators of ras [growth factors and receptor tyrosine kinase (RTK) ligands] and downstream effectors (MAP kinases/ERK pathways) were targeted as a means to increase tumor cell sensitivity to apoptosis-inducing agents[15,16]. Unfortunately, little is known regarding knockdown of ras in bladder cancer cell lines using siRNA, despite the major role of ras in bladder cancer progression and chemotherapy resistance[2,17]. The aim of this study, therefore, was to elucidate the effect of knockdown of ras protein on the apoptotic response of MMC-resistant T24 cell lines. Although H-ras is the predominant mutated ras isoform in TCCB and in T24 cells, the presence of K-ras and N-ras may provide alternative pathways for tumor cell recovery and acquisition of resistance. Therefore, in this study, ras interference with the three isoforms of ras was employed to ensure potency.
Methods
Cell culture
The human bladder cancer cell line T24, obtained from the European Collection of Authenticated Cell Cultures (ECACC), were grown in confluent monolayers with McCoy's 5A medium (Sigma-Aldrich Co. Ltd., Poole, Dorset, England) supplemented with 10% fetal calf serum (FCS), penicillin G (100 IU/mL), streptomycin (100 µg/mL), and L-glutamine (2 mmol/L) and were maintained at 37 °C with 5% CO2. Cells were grown on coverslips for subsequent morphology and immunocytochemistry. They were grown in T75 flasks for flow cytometry and Western blotting assessments.
Treatment protocols
MMC treatment: MMC cross links DNA, therefore, DNA damage is time- and concentration-dependent. In experiments examining the effect of MMC alone, MMC (Kyowa Hakko UK Ltd, England) containing media (0.1, 0.3, 1, 3, 10, 30, and 100 μg/mL) was applied to cells for 1h to simulate the microenvironment during intravesical therapy[18]. MMC containing media was then withdrawn and cells were washed and incubated with drug-free media to examine the delayed effects of MMC alone after 24 and 72 h.
Ras siRNA transfection: The siRNA used was a human pan-ras that targets all isoforms of ras. Human pan-ras siRNA and the non-targeting negative control siRNA were syntesized according to manufacturer's instructions (siIMPORTER transfection kit, Upstate Biotechnology, VA, USA). In experiments examining the effect of ras siRNA alone, cells were transfected with pan-ras siRNA at concentrations of 50 and 100 nmol/L and were incubated in serum-free medium for 4 h at 37 °C. FCS was then added and cells were incubated for 72 h. A full set of controls were used to detect false-negative or false-positive results.
Combination treatment: In experiments that investigated the combined effect of MMC and ras siRNA, T24 cells were exposed to low concentrations of MMC (0.1, 0.3, 1, 3, and 10 μg/mL) for 1 h, and then were washed and further transfected with 50 nmol/L of ras siRNA. The effect of MMC-ras siRNA combination was tested after 72 h.
Morphological assessment of apoptosis
Tumor cells grown on coverslips were fixed in 70% alcohol for 1 h and stained with Giemsa or hematoxylin and eosin. We assessed architectural features which described the overall arrangement of the sheets of growing cells and the extent of cell loss, mitotic activity, apoptotic count, and the most prevalent apoptotic stage on a set of three slides, using the 40×, 100×, and 400× objectives on a Leica DM2500 microscope (Leica, Germany) equipped with high resolution DC300 Leica camera. A combined morphological scoring system was devised as follows: (1) apoptotic count/100 cells: 0-5 = 1; 5-10 = 2; > 10 = 3; (2) phase of apoptosis: early apoptosis shown by nuclear shrinkage and formation of cell blebbing = 1; late apoptosis shown by formation of apoptotic bodies = 2; (2) mitotic count/100 cells: decreased by one third = 1; decreased by two thirds = 2; no mitosis = 3; and (4) architectural pattern: irregular small clefts/spaces = 1; spaces coalesced = 2; large spaces with few cells = 3. All assessments were repeated three times by three independent observers.
Flow cytometric analysis
Progression of cells through the cell cycle and cell apoptosis were measured by propidium iodide (PI)/fluorescence-activated cell analysis of subGo/G1 DNA content[19]. After fixation in 70% alcohol for 1 h, cells were washed and suspended in 1 mL fluorescent probe solution containing PBS, 1% Triton X-100, 50 µg/mL PI and 0.5 mg/mL RNase for 30 min in the dark at room temperature. Ten thousand events were acquired in a Coulter Epics Elite flow cytometer (Beckman Coulter, Florida, USA) equipped with a 488 nm argon laser. Cells were discriminated according to levels of red fluorescence emitted from the PI, which was collected using the 610 nm long band pass filter. DNA histograms were analyzed using WinCycle version 3.0 (Phoenix Flow Systems, San Diego, USA).
Immunocytochemistry
Alcohol-fixed cells were washed prior to immunostaining using 5 µg/mL anti-pan-ras (clone RAS10, Upstate, VA, USA), 1 µg/mL rabbit polyclonal ERK-1, which reacts with both ERK-1 and ERK-2 (K-23; Santa-Cruz Biotechnology, Santa-Cruz, CA), 1 µg/mL mouse anti human Ki-67 (Clone MIB-1, Dako, UK) or matched isotypes IgG2a (ras) and IgG1 (ERK and Ki-67) at equivalent concentrations diluted in PBS with 0.04% Tween 80 (PBST). Immunoreactivity was visualized using HRP anti-mouse IgG (DAB+) EnVision Kit (Dako) and the stained slides were analyzed by digital image analysis.
Digital image analysis
Computer-aided quantification of ICC staining was performed based on the principle originally described by Dahab et al.[20] with slight modification[21]. Briefly, digital images were acquired at 100× using a Leica digital camera and were analyzed with VideoTesT-Morphology software (Russia). For ras and ERK, color sampling was selected and the number of selected pixels was read from the histogram of colors. The percentage per section was determined. We estimated Ki-67 labelling indices (LIs) reflecting the proliferation activity of tumor cells according to a recently described method[22]. The Ki-67 LI was calculated as 100 × Ki-67 positive nuclei/(Ki-67-positive nuclei + Ki-67-negative nuclei) per image. Ten random images from each slide were analyzed and averaged.
Western blotting
5 × 106 cells were lysed for 15 min on ice using 300 µL of CelLytic-M Mammalian Reagent (Sigma-Aldrich, UK). Protein concentration in the supernatant was determined using the BCA Protein Assay Kit (Pierce, USA). Lysates were diluted in NuPAGE 4X LDS sample buffer (Invitrogen USA). Ten micrograms of the total protein were denatured, reduced, and resolved on NuPAGE Novex 4%-12% Bis-Tris Gels (Invitrogen, USA) by SDS-PAGE prior to transfer onto 0.2 µmol/L pore size nitrocellulose membrane. After incubation in blocking solution [1% bovine serum albumin (BSA) in Tween-20/TBS (TBST)] for 2 h at room temperature, membranes were incubated with 1 µg/mL anti-pan-ras or ERK antibodies diluted in 5 mL of TBST overnight at room temperature.
Statistical analysis
Statistical analysis was performed using SPSS 15.0 (SPSS Inc, Chicago, IL, USA). Data were quantified from triplicate samples and were presented as the mean ± SD. Multiple comparisons between means was performed using one-way ANOVA followed by Dunnett's post hoc test. A P-value of less than 0.05 was considered statistically significant.
Results
T24 cells sensitivity after MMC treatment alone
Untreated T24 cells grew in irregularly dispersed sheets comprising cells of variable sizes and shapes [Figure 1]. Apoptotic cells were only very occasionally identified, usually in the early apoptotic phase. Apoptotic bodies were observed 24 h after treatment with very low doses of MMC. The apoptotic count score was 1 after 1.0, 3.0, and 10 µg/mL, and increased to 2 after 30 and 100 µg/mL [Figure 2]. By contrast, the mitotic count decreased significantly at 10, 30 and 100 μg/mL, and this was reflected as increases in the mitotic score. Architectural differences were observed after treatment with MMC 30 µg/mL (score = 1) and were more pronounced after treatment with 100 µg/mL (score = 2). There was a dose-dependent increase of subGo/G1 events as shown by DNA flow cytometry [Figure 3]. The intensity of ras and ERK immunoreactivity in T24 cells significantly increased 24 h after treatment with MMC doses ≥ 10 μg/mL, compared with that of untreated cells. This suggests enhanced ras protein synthesis countering the anti-proliferative effects of MMC (P < 0.001 for all). This was also confirmed by Western blot analysis of ras protein expression [Figures 4-6]. The percentage of Ki-67 labelled cells decreased progressively with increasing doses of MMC from 75.0% ± 7.0% without treatment to 15% ± 2.4% at 100 μg/mL (P < 0.001, Figure 7).
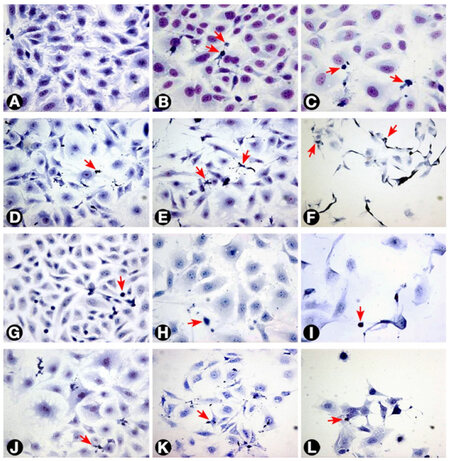
Figure 1. Morphology of T24 cells as seen with different treatments. (A) Untreated cells; (B and C) cells treated with 3 and 30 μg/mL MMC respectively after 24 h; (D) cells treated with 0.3 μg/mL MMC after 72 h; (E and F) cells treated with 3 and 30 μg/mL MMC after 72 h; (G) cells treated with negative control ras siRNA after 72 h; (H and I) cells transfected with 50 and 100 nmol/L ras siRNA after 72 h, respectively. The cells became larger with loss of spindling. The architecture was significantly disrupted with marked decrease in cellularity; (J, K and L) cells treated with 0.3, 3, and 10 μg/mL MMC, respectively, combined with 50 nmol/L ras siRNA after 72 h. The arrows indicate apoptotic cells (Giemsa 400×). MMC: mitomycin C
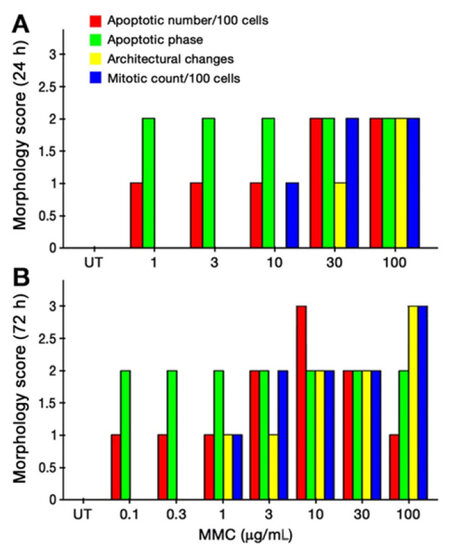
Figure 2. Combined morphological score of T24 cells after 24 h (A) and 72 h (B) of MMC treatment. MMC: mitomycin C; UT: untreated cells
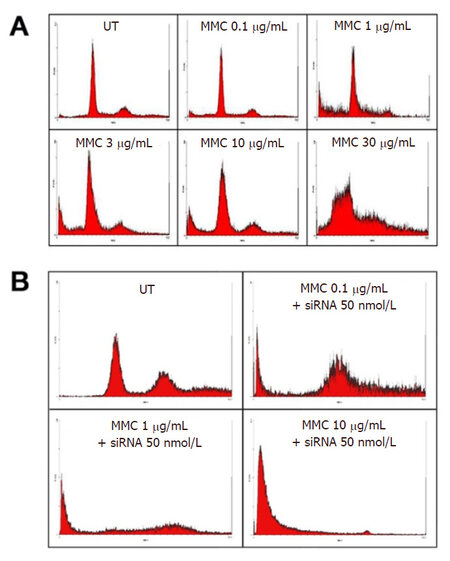
Figure 3. SubGo/G1events in T24 cells using DNA flow cytometry after 72 h of incubation with different concentrations of MMC alone (A) or in combination with ras siRNA (B). The DNA histograms illustrate an increase in percentage of SubGo/G1events at 30 and 100 μg/mL MMC. Combination with 50 nmol/L ras siRNA increased subGo/G1 events when compared with MMC alone. MMC: mitomycin C; UT: untreated cells
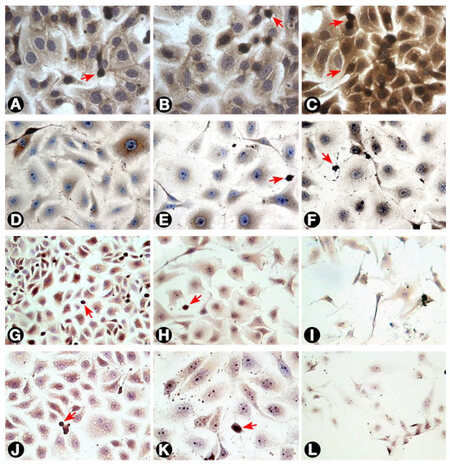
Figure 4. Ras expression by T24 cells as visualized by immunostaining. (A) Untreated cells; (B and C) T24 cells treated with 3 and 30 μg/mL MMC after 24 h. The intensity of staining increased after treatment as compared with untreated cells; (D, E, and F) cells treated with 0.3, 3, and 30 μg/mL MMC after 72 h, respectively. The staining intensity showed progressive reduction with increasing MMC dose to reach the minimum after treatment with 100 μg/mL (not shown); (G, H, and I) cells treated with negative control, 50, and 100 nmol/L ras siRNA after 72 h, respectively. The cells showed little intensity of staining when compared to negative control with the least intensity at 100 nmol/L; (J and K) cells treated with 0.3 and 3 μg/mL MMC, respectively, combined with 50 nmol/L ras siRNA and cultured for 72 h; (L) cells treated with 3 μg/mL MMC combined with 100 nmol/L ras siRNA and cultured for 72 h. The cells undergoing apoptosis or mitosis (arrows) were more intensely stained compared with other cells (400×). MMC: mitomycin C
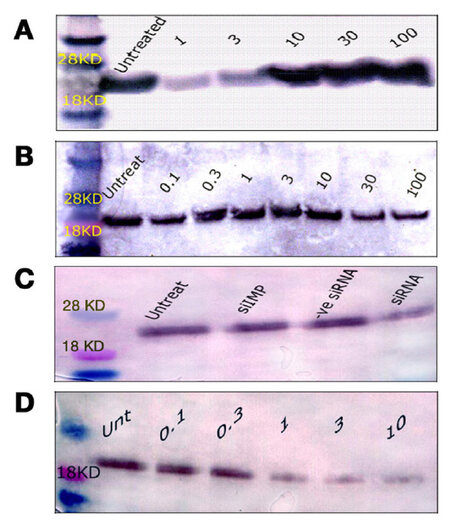
Figure 5. Western blotting of ras protein expression in T24 cells. (A) ras protein expression 24 h after treatment with 1, 3, 10, 30, or 100 μg/mL MMC; (B) ras protein expression 72 h after treatment with 0.1, 0.3, 1, 3, 10, 30, or 100 μg/mL MMC. There was a progressive decrease of the blot thickness and intensity with increasing doses of MMC; (C) the level of ras following 50 nmol/L ras siRNA was low compared with untreated cells, or with other controls; (D) ras protein changes 72 h after low doses of MMC combined with 50 nmol/L of ras siRNA. All bands appeared between 18 and 28 kDa at the standard ras molecular weight of 21 kDa when compared with the molecular weight marker. MMC: mitomycin C
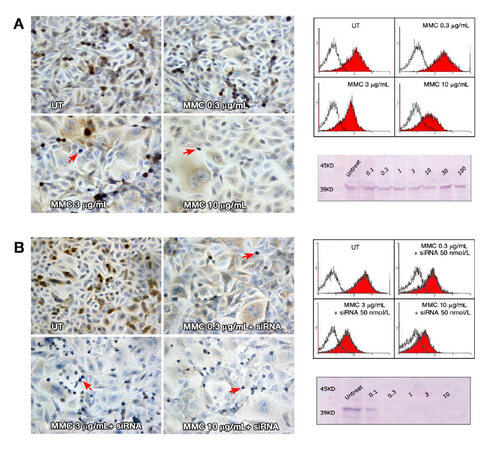
Figure 6. (A) ERK expression in T24 cells 72 h after MMC treatment alone. ERK staining showed progressive decline in the staining intensity with increasing MMC dose; (B) ERK staining with MMC combined with siRNA, where the decline in staining intensity was greater when MMC was combined with ras siRNA. In all slides, the apoptotic and mitotic cells (arrows) were negative for ERK staining (200×). To the right, overlay histograms of flow cytometry show the extent of ERK specific fluorescence (red-filled) in comparison to isotype control fluorescence (unfilled). Western blot traces show mild decrease of the blot thickness and intensity at 10, 30, and 100 μg/mL MMC compared to the UT control (above), with nearly complete absence of the blot when MMC was combined with ras siRNA (below). All the bands appeared between 39 and 45 kDa on the molecular marker at the standard ERK 1/2 molecular weight of 42/44 kDa. MMC: mitomycin C; UT: untreated cells
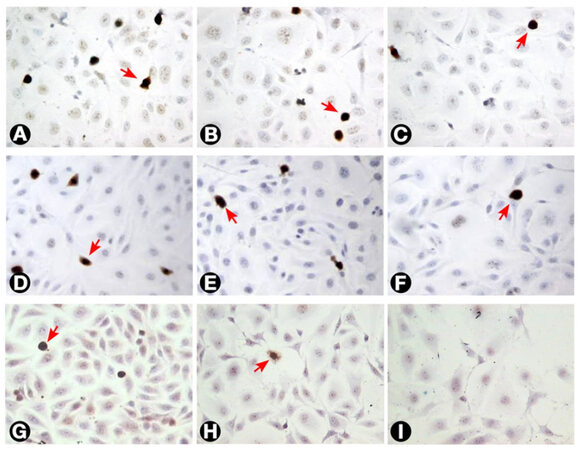
Figure 7. The incidence of Ki-67 labelled T24 cells as seen with different treatments. (A) Untreated cells; (B and C) cells treated with 3 and 10 μg/mL MMC, respectively, after 24 h; (D, E, and F) cells treated with 0.3, 3, and 10 μg/mL MMC, respectively after 72 h. The percentage of Ki-67 labelled cells (arrows) decreased progressively with increasing dose and duration of MMC treatment; (G, H, and I) cells treated with 0.3, 3, and 10 μg/mL MMC combined with 50 nmol/L ras siRNA after 72 h. The decrease in Ki-67 positivity was higher than with MMC alone (200×). MMC: mitomycin C
After 72 h, there were isolated strands randomly dispersed amongst large spaces at doses ≥ 3 µg/mL MMC [Figure 1]. The apoptotic count score was 1 after 0.1, 0.3, and 1.0 µg/mL. It reached a maximum of 3 after 10 µg/mL and, gradually began to decline at doses greater than 10 µg/mL [Figure 2]. Following treatment with 10, 30, and 100 µg/mL, mitotic count progressively decreased (score = 1, 2, and 3, respectively). There was substantial architectural deterioration (score = 3) at 100 µg/mL MMC. The amount of apoptosis, as assessed by DNA flow cytometry, was higher at 72 h than at the 24 h time point. Treatment with 1 µg/mL MMC induced a 1.9-fold increase in subGo/G1 events (P < 0.005), while treatment with 10 µg/mL induced a 3.8-fold increase in subGo/G1 events (mean subGo/G1 = 22.2 ± 3.9 compared with 5.8 ± 0.6 in untreated cells, P < 0.001, Figure 3). The staining intensity of ras and ERK decreased progressively from 22.7% ± 3.5% and 17.2% ± 3.1% in untreated cells, to 13.5% ± 2.2% and 9.4% ± 2.0%, respectively, after treatment with 10 µg/mL MMC (P < 0.001 for all), to nearly complete absence of staining of both proteins after treatment with 100 µg/mL MMC. These findings were confirmed using flow cytometry and Western blotting [Figures 4-6]. The percentage of Ki-67 labelled cells decreased significantly with increasing the dose and duration of MMC compared with that of untreated cells [Figure 7].
T24 cell sensitivity after ras siRNA treatment alone
ras siRNA treated cells were compared with control slides for better characterization of the effect of ras knockdown on the T24 cell phenotype. After 72 h of transfection with 50 nmol/L ras siRNA, T24 cells showed loss of spindling and substantial architectural disruption [Figure 1]. This was associated with increase in subGo/G1 events by 1.1-fold (P < 0.01 vs. untreated). Ras immunoreactivity decreased but ERK immunoreactivity increased. By contrast, 100 nmol/L of ras siRNA increased subGo/G1 events by 1.8-fold (P < 0.001 vs. untreated) and decreased both ras and ERK immunoreactivity [Figure 4]. These data were further supported by flow cytometry and Western blotting. The mean of Ki-67 labelled cells also decreased after transfection with 50 and 100 nmol/L ras siRNA (45.5% ± 5.4% and 36.2% ± 4.8%, respectively, compared with 72.0% ± 6.8% in control cells, P < 0.001 for both). Although there were few observed changes in the measured parameters following the use of negative control ras siRNA compared with the untreated cells, these changes were not significant as compared with the effects induced by ras siRNA at 50 and 100 nmol/L concentrations [Figure 4].
T24 cell sensitivity after combined MMC and ras siRNA treatment
Because of the significant cell death induced in T24 cells following the individual use of high concentrations of either MMC or ras siRNA observed in our pilot study, we used lower doses of MMC (maximal 10 µg/mL) in combination with low-dose ras siRNA (50 nmol/L) in the synergy experiment, and the results were assessed after incubation for 72 h of ras siRNA transfection. The morphological pattern of T24 cells treated with low concentrations of MMC in combination with 50 nmol/L ras siRNA was similar to that of cells treated with ten-fold high concentration of MMC alone [Figure 1]. In addition, the combined treatment with 10 µg/mL MMC and 50 nmol/L ras siRNA induced three times as many subGo/G1 events compared to the number induced by l0 µg/mL MMC alone [Figure 3]. The decrease in the intensity of ras and ERK immunoreactivity visualized after combined treatment was greater than that observed after either MMC or ras siRNA were used alone [Figures 4 and 6]. The staining values for ras and ERK proteins were significantly correlated (r = 0.867, Pearson correlation, data not shown). Significant synergistic effects were observed when MMC concentrations exceeded 1 µg/mL. The percentage of Ki-67-positive cells decreased progressively to reach the lowest value at 3 and 10 μg/mL of MMC. The decrease in Ki-67-positivity was greater than when either MMC or ras siRNA were used alone [Figure 7].
Discussion
Therapy for bladder cancer remains associated with persistent high rates of failure resulting from the acquisition of chemoresistance. Therefore, novel therapeutic combinations and treatments are required to improve chemoresponsiveness. In this study, we investigated the synergistic antitumor effects of low doses of MMC and pan-ras siRNA in T24 bladder cancer cells that had previously been reported to be MMC-resistant[23]. We reasoned that the tumorigenic role of ras in T24 cells may provide key information to our understanding as to why 30% to 60% of bladder tumors that carry mutated ras have a propensity to induce tumor aggressiveness and resistance[17,24]. This direction was further suggested by the fact that T24 bladder cancer cells represent a wide variety of tumors carrying mutant ras protein that are clinically linked with occupational and environmental bladder cancers[25].
Finding a synergy between drug combinations should be examined carefully. The synergism should include augmentation of the action of one drug by another drug that has a different mechanism of action in contact with a common target. The common objective of MMC and siRNA synergy experiments is to break MMC resistance of T24 cells (induction of higher apoptosis level) through inhibition of ras-induced resistance. We tested the combination of MMC and ras siRNA for possible synergy using low doses of MMC and a fixed dose of 50 nmol/L of siRNA that was determined from pilot experiments (data not shown) to be the most suitable for combination with MMC. In addition, the understanding of the proper MMC dose/time point was acquired from preliminary experiments which suggested that MMC treatment using low concentrations over 72 h was the best combination regimen with ras siRNA. This pre-rationale assessment strategy meant that the siRNA study design was based on a solid background and knowledge of how the proteins behaved in response to single agents. This was extremely important because the “hit” of the second therapy could be timed to coincide with the lowest expression of the target.
We showed that T24 cells express more ras, supporting previous reports that ras expression was associated with tumor cell resistance[24,26]. We observed increased ras immunoreactivity in T24 cells 24 h after MMC treatment. This could be attributed to enhanced ras protein synthesis to counter the anti-proliferative effects of MMC. Previous reports have shown that expression of ras enhances pro-survival signaling pathways after DNA damage induced by chemotherapeutic agents[27,28]. Contrary to this effect, we observed ras immunoreactivity was decreased 72 h after treatment with low concentrations of MMC. This could be attributed to depletion of the protein synthesis capacity in T24 cells after 72 h[29]. These data suggest that ras siRNA would be most effective 72 h after treatment with low concentrations of MMC.
ERK is the direct downstream effector pathway responsible for cell resistance, and hence acts as a surrogate marker of ras activation status and expression levels[30]. To explore further the effects of ras siRNA alone and in combination with MMC, we designed a set of experiments employing the anti-ERK1/ERK2 antibody. Several studies have shown that ras activates a number of signal transduction pathways, including the RAF-MAP2K-MAP kinase pathway[16,31]. MAP kinases including ERK1/ERK2 have also been shown to be activated following radiation through this pathway. Activation was dependent on mutant ras in T24 cells, while wild type ras, which is induced by radiation in RT4 cells, was independent of ras[32]. This substantial body of literature would suggest that ERK plays a critical key role downstream of ras activation[9,13]. In our study, low concentrations of MMC, 100 nmol/L ras siRNA, and combination therapies decreased ERK immunoreactivity and were associated with increased apoptosis, while 50 nmol/L ras siRNA increased ERK immunoreactivity and was associated with only marginal apoptosis. On the other hand, the increase in ERK after 50 nmol/L ras siRNA may be explained by Mishra et al.[33], who identified the presence of a ras-independent pathway for ERK stimulation through the Rap-1 cascade of MAP kinase activation.
We speculate that a small reduction in ras protein levels could be compensated for by ras-independent pathways, but that significant decreases in combination with inhibition of ras synthesis would not be compatible with compensatory mechanisms. Hence the combination of ras siRNA and MMC was synergistic and achieved the greatest decreases in ras synthesis and ERK expression. This combination of withdrawal of survival factors and induction of apoptosis achieved a synergistic combination through targeting different pathways of apoptotic resistance[34]. Interconnectivity of oncogenic signal transduction networks leads to drug resistance. This underscores the need to investigate combination therapies which inhibit two pathways or which are designed to hit one pathway and its corresponding feedback loop[35].
In conclusion, the data presented in this study suggest that T24 cell resistance to MMC-induced apoptosis is dependent on ras activation. We demonstrated that ras siRNA knockdown in combination with low dose MMC was synergistic in T24 cells at specific pre-determined optimal timings. Translation of this therapeutic strategy may be feasible following validation of in vivo timing to ensure that ras siRNA administration coincides with optimal MMC effects.
Declarations
Authors’ contributionsConceptualization, methodology, software, formal analysis, investigation, resources, data analysis, writing original draft, review and editing, visualization, and project administration: Sharaf Eldin O
Conceptualization, methodology, formal analysis, investigation, resources, data curation, writing original draft, and revision of the final version: Fouda AM
Conceptualization, methodology, validation, investigation, data analysis, and revision: Youssef AR
Conceptualization, methodology, validation, investigation, resources, review and editing, visualization, supervision, and project administration: Hamilton P
Investigation, resources, revision: Maxwell P
Conceptualization, methodology, validation, formal analysis, investigation, resources, data analysis, writing original draft and editing, visualization, supervision, and project administration: Williamson KE
Data source and availabilityCorresponding author may be contacted for any data inquiries.
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalThis research study was approved by the institutional review board of the study site.
Copyright© The Author(s) 2018.
REFERENCES
1. Niino M, Matsuda T. Morphological distribution of bladder cancer from Cancer Incidence in Five Continents Vol. X. Jpn J Clin Oncol 2015;45:999.
2. Hurst CD, Alder O, Platt FM, Droop AD, Stead LF, Burns JE, Burghel GJ, Jain S, Klimczak LJ, Lindsay H, Roulson J, Taylor CF, Thygesen H, Cameron AJ, Ridley AJ, Mott HR, Gordenin DA, Knowles MA. The genomic landscape of non-muscle-invasive bladder cancer: implications for molecular classification and treatment. Cancer Res 2016;76:LB-323.
3. Bryan RT. Optimising existing therapeutic strategies for the treatment of non-muscle-invasive bladder cancer: the role of intensive neoadjuvant intravesical mitomycin C. Eur Urol 2012;62:803-5.
4. Birare N, Lwaleed BA, Cooper AJ. Multidrug resistance in a urothelial cancer cell line after 1-hour mitomycin C exposure. J Urol 2009;182:2472-6.
5. Xu B, Sun Y, Singh SV. Mechanism of resistance to mitomycin C in a human bladder cancer cell line. Zhonghua Zhong Liu Za Zhi 1995;17:343-6.
6. Sylvester RJ, Brausi MA, Kirkels WJ, Hoeltl W, Calais Da Silva F, Powell PH, Prescott S, Kirkali Z, van de Beek C, d Gorlia T, de Reijke TM; EORTC Genito-Urinary Tract Cancer Group. Long-term efficacy results of EORTC genito-urinary group randomized phase 3 study 30911 comparing intravesical instillations of epirubicin, bacillus Calmette-Guerin, and bacillus Calmette-Guerin plus isoniazid in patients with intermediate- and high-risk stage Ta T1 urothelial carcinoma of the bladder. Eur Urol 2010;57:766-73.
7. Shinohara N, Koyanagi T. Ras signal transduction in carcinogenesis and progression of bladder cancer: molecular target for treatment? Urol Res 2002;30:273-81.
8. Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 1982;297:474-8.
9. Maertens O, Cichowski K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul 2014;55:1-14.
10. Boulalas I, Zaravinos A, Karyotis I, Delakas D, Spandidos DA. Activation of RAS family genes in urothelial carcinoma. J Urol 2009;181:2312-9.
11. Johne A, Roots I, Brockmöller J. A single nucleotide polymorphism in the human H-ras proto-oncogene determines the risk of urinary bladder cancer. Cancer Epidemiol Biomarkers Prev 2003;12:68-70.
12. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002;298:1911-2.
13. Handra-Luca A, Bilal H, Bertrand JC, Fouret P. Extra-cellular signal-regulated ERK-1/ERK-2 pathway activation in human salivary gland mucoepidermoid carcinoma. Association to aggressive tumor behavior and tumor cell proliferation. Am J Pathol 2003;163:957-67.
14. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Dis 2014;13:928-42.
15. Kim J, Akbani R, Creighton CJ, Lerner SP, Weinstein JN, Getz G, Kwiatkowski DJ. Invasive bladder cancer: genomic insights and therapeutic promise. Clin Cancer Res 2015;21:4514-24.
16. Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 2003;17:1263-93.
17. Oxford G, Theodorescu D. The role of Ras superfamily proteins in bladder cancer progression. J Urol 2003;170:1987-93.
18. De Bruijn EA, Sleeboom HP, van Helsdingen PJ, van Oosterom AT, Tjaden UR, Maes RA. Pharmacodynamics and pharmacokinetics of intravesical mitomycin C upon different dwelling times. Int J Cancer 1992;51:359-64.
19. Crowley LC, Scott AP, Marfell BJ, Boughaba JA, Chojnowski G, Waterhouse NJ. Measuring cell death by propidium iodide uptake and flow cytometry. Cold Spring Harb Protoc 2016;2016:pdb.prot087163.
20. Dahab GM, Kheriza M, El-Beltagi HM, Fouda AM, Sharaf El-Din O. Digital quantification of fibrosis in liver biopsy sections: description of a new method by Photoshop software. J Gastroenterol Hepatol 2004;19:78-85.
21. Rizzardi AE, Johnson AT, Vogel RI, Pambuccian SE, Henriksen J, Skubitz AP, Metzger GJ, Schmechel SC. Quantitative comparison of immunohistochemical staining measured by digital image analysis versus pathologist visual scoring. Diagn Pathol 2012;7:42.
22. Laurinavicius A, Plancoulaine B, Laurinaviciene A, Herlin P, Meskauskas R, Baltrusaityte I, Besusparis J, Dasevicius D, Elie N, Iqbal Y, Bor C. A methodology to ensure and improve accuracy of Ki67 labelling index estimation by automated digital image analysis in breast cancer tissue. Breast Cancer Res 2014;16:R35.
23. Vasquez JL, Gehl J, Hermann GG. Electroporation enhances mitomycin C cytotoxicity on T24 bladder cancer cell line: a potential improvement of intravesical chemotherapy in bladder cancer. Bioelectrochemistry 2012;88:127-33.
24. Wood Jr DP, Anderson AE, Fair R, Chaganti RS. Ras oncogene point mutations in bladder cancer resistant to cisplatin. Urol Res 1992;20:313-6.
25. Ou L, Guo Y, Luo C, Wu X, Zhao Y, Cai X. RNA interference suppressing PLCE1 gene expression decreases invasive power of human bladder cancer T24 cell line. Cancer Genet Cytogenet 2010;200:110-9.
26. Stanslas J, Wong CC, Sagineedu SR, Sidik S, Sumon SH, Phillips R, Lajis NH. Mechanism of resistance to a semisynthetic anticancer andrographolide derivative: possible involvement of Ras-MAPK signaling pathway. Mol Cancer Ther 2013;12:C147.
27. Cho HJ, Jeong HG, Lee JS, Woo ER, Hyun JW, Chung MH, You HJ. Oncogenic H-Ras enhances DNA repair through the Ras/Phosphatidylinositol 3-Kinase/Rac1 pathway in NIH3T3 cells. Evidence for association with reactive oxygen species. J Biol Chem 2002;277:19358-66.
28. Balko JM, Denkert C, Salgado R, O'Hely M, Savas P, Beavis PA, Darcy PK, Combs S, Rimm DL, Giltnane JM, Estrada MV, Sanders ME, Cook RS, Wang K, Miller VA, Stephens PJ, Yelensky R, Pinto JA, Doimi F, Gomez H, Arteaga CL, Loi S. Reduced tumor lymphocytic infiltration in the residual disease (RD) of post-neoadjuvant chemotherapy (NAC) triple-negative breast cancers (TNBC) is associated with Ras/MAPK activation and poorer survival. Cancer Res 2015;75:S1-8.
29. Matzno S, Yasuda S, Juman S, Yamamoto Y, Nagareya-Ishida N, Tazuya-Murayama K, Nakabayashi T, Matsuyama K. Statin-induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J Pharm Pharmacol 2005;57:1475-84.
30. Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J 2013;280:489-504.
31. Nan X, Tamgüney T, Collisson EA, Lin L, Pitt C, Galeas J, Lewis S, Gray J, McCormick F, Chu S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. PNAS 2015;112:7996-8001.
32. Gupta AK, Bernhard EJ, Bakanauskas VJ, Wu J, Muschel RJ, McKenna WG. RAS-Mediated radiation resistance is not linked to MAP kinase activation in two bladder carcinoma cell lines. Radiat Res 2000;154:64-72.
33. Mishra S, Smolik SM, Forte MA, Stork PJ. Ras-independent activation of ERK signaling via the torso receptor tyrosine kinase is mediated by Rap1. Curr Biol 2005;15:366-70.
34. Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, Wick MJ, Alvarez C, Cavazos A, Mangold GL, Patnaik A. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 2012;18:2316-25.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Eldin OS, Fouda AM, Youssef AR, Hamilton P, Maxwell P, Williamson KE. Reduction of mitomycin C resistance in human bladder cancer T24 cells by knocking-down ras oncogene. Cancer Drug Resist 2018;1:59-71. http://dx.doi.org/10.20517/cdr.2017.01
AMA Style
Eldin OS, Fouda AM, Youssef AR, Hamilton P, Maxwell P, Williamson KE. Reduction of mitomycin C resistance in human bladder cancer T24 cells by knocking-down ras oncogene. Cancer Drug Resistance. 2018; 1(1): 59-71. http://dx.doi.org/10.20517/cdr.2017.01
Chicago/Turabian Style
Eldin, Osama Sharaf, Abdel-Motaal Fouda, Amany R. Youssef, Peter Hamilton, Perry Maxwell, Kate E. Williamson. 2018. "Reduction of mitomycin C resistance in human bladder cancer T24 cells by knocking-down ras oncogene" Cancer Drug Resistance. 1, no.1: 59-71. http://dx.doi.org/10.20517/cdr.2017.01
ACS Style
Eldin, OS.; Fouda A.M.; Youssef AR.; Hamilton P.; Maxwell P.; Williamson KE. Reduction of mitomycin C resistance in human bladder cancer T24 cells by knocking-down ras oncogene. Cancer Drug Resist. 2018, 1, 59-71. http://dx.doi.org/10.20517/cdr.2017.01
About This Article
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 0
likes
Like This Article 0
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.